在药品的开发和最终商业化过程中必须指定有效期。有效期使用各种因素来确定在合理的储存条件下产品对病人安全有效的时限。有效期不仅作为临床试验申请提交的材料也应用于商业化产品。在某些情况下,会指定两个有效期:一个用于原始包装的散装剂型,另一个对应于从初级包装中取出制剂后的安全储存时间,这对于需给病患用药前需配制的药品尤其重要。
1、化学稳定性
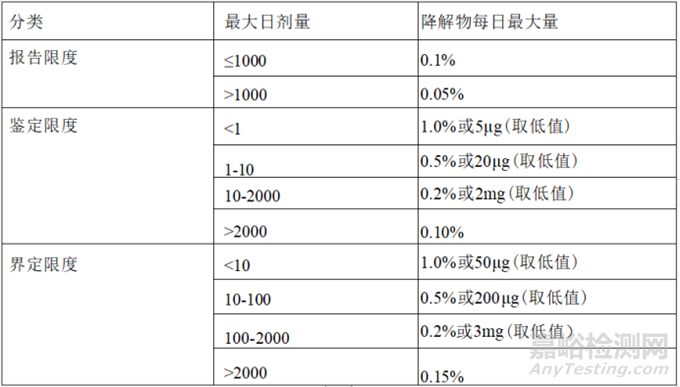
药物活性成分(APIs),无论是生物成分(即蛋白质还是核酸)或小分子,容易受到有机化学降解的影响,为了保证药品的安全性和有效性,监管机构要求对降解产物进行评估以指定有效期。药品的有效期是根据在规定的可能贮藏条件下发生某些变化的时间所制定,这些变化包括降解物水平达到影响安全性或活性成分的含量或活性降至临界水平以下所需的时间。降解物需进行限度规定,降解物限度可根据摄入API的总日剂量计算,并需进行相应报告,鉴别和定量限度。报告限度为降解物存在的限度,达到该限度必须向监管机构报告以提醒他们降解物的含量,通常可在HPLC检出峰的存在。鉴定限度定义为一限度,大于该限度的降解产物需鉴定其结构。最后,界定限度为需进行毒理学评估以确保安全性评价的限度。这些限度定义为API每日总摄入量的百分比或绝对质量,取数值较低者。下表说明了特定降解物的不同限度要求,该要求与ICH 的建议一致。各阈值通常允许的特定降解产物水平。
用于确定特定降解产物水平的容许因素取决于以下因素:
1)如果降解物也是一种代谢物,那么降解物水平的限度允许较高,因为该类降解物在API的临床研究中已被证实其安全性。
2)若原料药处于低剂量时,在可接受的暴露量下,其降解物的比例接受上限可稍微高点。
3)若降解物是可能为致癌物、致畸剂或致突变,则应采用更低的限度。
4)若要长期服用该API,允许的降解物限度可能低于单次给药下降解物限度。
5)在有效期末,缺乏安全信息的降解物一般定义其限度为在API的0.2-0.5%。
值得注意的是,在某些情况下,降解物也可以是工艺杂质(如,存在于初始未处理的API 样品中)。稳定性限度反映了物料的总量,包括最初存在的数量和在储存条件下形成的数量。
在某些情况下,已知降解物是无害的(如,药物前体的降解)。在这些情况下,有效期的限制因素将是API含量的下降。虽然这对患者不会造成直接的安全风险,但如果患者没有接受预期剂量的原料药仍可能造成危害。一般来说,基于ICH指导原则,效期末的含量需要保持在标示量的95%或以上。若考虑到初始含量的可变性,则影响更为显著。
大分子药物如核酸、蛋白质和寡核苷酸、化学降解部分分子与活性位点无关,对生物活性可能没有显著影响。正因为如此,尽管化学降解量可能相当显著也不至于限制有效期。活性通常是用生物效力指示实际的活度。当化学降解发生在生物分子的活性位点附近,其活性损失可能与小分子一样显著;然而,若降解并不直接或通过二级或三级结构改变影响活性位点,即使蛋白质的侧链降解40%也不会影响它的活性。
在确定药品的有效时,最具限制性的因素将成为决定型因素。例如,如果基于毒性问题某单一降解产物具有非常严格的限度,即使迅速形成不同的降解产物,其也可能是设定有效期的限制因素。一般来说,在存储条件下,即使在效期末,制剂产品中原料药转化为降解物的比例也相对较低。因此,没必要在制剂产品的降解过程中全程监测此项。实际上,达到有效期后限制因素发生的情况变得无关紧要。该论述可用于加速试验中,并将在后续章节讨论。
对于由药剂师或患者配制的液体制剂,在配制前和配制后应单独注明其有效期。配制前的物理稳定性意味着配制前制剂的效期应该涵盖配制后如溶解或分散的稳定性。配制后,稳定性问题与其他液体制剂类似。即使是固体剂型,药品运输(可能带有更加保护性的包装)和患者接收时的有效期也不同。
2、物理稳定性
在某些情况下,有效期的可能是受到制剂物理稳定性而不是其化学稳定性限制。物理稳定性最重要的是在储存后引起剂型性能的改变。其中特别关注的是可能改变其API生物利用度的任何因素。对于固体剂型,这可能意味着溶解性能发生了变化(崩解和随后的溶解)。片剂和胶囊剂在储存时出现溶出度变化的原因有很多。对于片剂,大多数问题与从环境中吸收水分有关,这使得崩解剂的效力改变。崩解剂能够吸水迅速膨胀,使药片在胃中破裂。在储存过程中湿气被缓慢吸收,发生缓慢的膨胀,这阻碍了崩解剂在胃中快速崩解所需的爆炸性膨胀。当片剂暴露于胃液(或溶出杯)中时,崩解剂未能像药片在干燥时那样吸收更多的水,因此无法有效崩解。当崩解剂暴露于水冷凝的环境下,这样的问题尤其显著,如运输时药品从初始包装的气候带经过更冷的气候带,此时片剂从较高的温度和湿度转至低于此前露点。可能发生这种情况。
对于明胶胶囊(包括普通胶囊和软胶囊),胶囊本身在长期保存后容易发生物理变化。在某些情况下,由于在制剂或包装中的杂质水平较低,明胶将发生交联反应。此类反应通常是由小分子醛(例如甲醛和乙二醛)引起的。这种交联可能使得明胶在标准溶出介质中的溶解变得缓慢。但应注意在许多情况下,由于酶促过程的存在,体内释放不会受交联反应的影响。该影响可采用含有适当消化酶的特定溶出介质进行监测。
固体制剂的另一潜在物理变化涉及API的晶型。大多数药物剂型采用的API具有独特的晶型。在大多数情况下,分子排列不是唯一的,也就是说,不同晶型的API具有不同能量。若制剂产品使用的API呈现多晶型且为高能形式,在储存期间则有可能出现晶型的改变。在极少数情况下,这种晶型的改变可能会改变生物利用度。这可能是由于溶解度的变化(稳定的多晶型物通常溶解度较低),溶出速率也相应被改变。即使使用是热力学最稳定的晶型,溶剂化或去溶剂化仍然可能随放置时间而发生。去溶剂化产生是溶剂分子 (包括水合物)从晶格中丢失。溶剂化则通常是向晶格中添加水。一个晶型变化的极端例子,如去溶剂化导致完全失去晶格,而使得API形成了无定型。这种晶型的改变通常提高了药物溶解度(溶出速率),但却降低了药物稳定性。
对于某些API, 制剂可利用其无定型或其他高能型态增加溶解度,从而提高生物利用度,此类剂型中的原料药具有自发结晶的潜力,因为该过程是放热的并且通常是自催化的(即一旦晶核产生,它们可以增加进一步结晶的速率)。尽管如此,通过添加合适的稳定剂,无定型系统可限制晶体生长,从而在较长的放置期间保持稳定。由于这种随时间的变化可能会导致API药效下降,有效期可能受此因素限制。
对于液体剂型,在储存时发生生物利用度改变通常表现为API或其他制剂组分的沉淀。可能导致沉淀的因素有很多。对于小分子,沉淀可能是由于溶液(悬浮液)的pH值发生变化。这种变化可能是由于吸收二氧化碳,某种成分发生化学降解例如由于氧化产生酸或碱或缓冲成分的损失。小分子化合物沉淀的另一个原因则是由于API粒径增加。这种效应称为奥斯特瓦尔德(Ostwald)熟化,由于较小的120K.C.Waterman晶体在逐渐溶解过程中,因自身能量低于较大的晶体,也同时在生长(较大的晶体具有较低的表面体积比,晶体表面很少本身具有更高能量的分子)。后续章节将进一步讨论了原料药和制剂产品物理稳定性。
对于生物分子,悬浮液的特性将取决于分子的二级和三级结构特征,这又涉及离子,范德华力和氢键合力。通常,大分子有多种构型它们通常具有相似的能量,但凝集趋势却大不相同,从而导致不同的生物活性。虽然最初形成的生物API的构型可能是单体且悬浮良好,但随放置时间的推移,大分子可能变性,从而导致聚集和沉淀。由于聚集状态无法与助悬剂重新平衡,根据LeChatlier的原理,这一过程可使沉淀加剧。更多详细信息请参见后续章节。
3、外观
有时,制备成制剂的API可能出现外观的改变,其物理稳定性或化学降解几乎没有变化。颜色变化这种化学变化可以通过肉眼敏感地监测出。尽管这种细微的化学变化不太可能对病人造成威胁,但外观变化可能会影响病人使用,因此这也会限制药品的有效期。
在干燥条件下(例如使用干燥剂)储存时,明胶胶囊可能会变硬。虽然脆性可能不会导致生物活性发生变化,但仍然可能产生不可接受的外观。
对于包衣有膜包衣的片剂,在某些情况下,片芯水分溶胀可能引起包衣破裂。虽然这样做也不应影响性能,但仍会干扰患者,因此将限制有效期(或需要特殊包装)。
4、微生物生长
对于注射剂,有效期的限制因素应关注微生物杂质。对于许多此类制剂,通常加入生物稳定剂添加以防止或至少减慢微生物的生长。一旦稳定剂被消耗,微生物杂质可能增长。对于其他处方,在放置过程中,包装的完整性可能决定微生物的生长趋势。
5、光化学降解
药品的光敏性会影响其有效期,在许多情况下也决定了产品的包装要求。在某些情况下,API可能吸收光照后引发化学反应并诱发化学降解。光化学反应通常包括氧化和自由基重排。间接光化学过程也是可能的,在这些情况下,光被处方中的某种物质吸收,而非API, 然后导致与API发生反应。最有问题的环境光波长是长紫外光和短可见光(蓝色)光,部分原因是由于能量(高能波长),也有可能是因为吸收光谱的重叠。大多数影响API稳定性的光过程以互为倒数的方式响应光强度,即当光的总通量是相同的,则短时间、高强度曝光与长时间、低强度曝光具有相同的影响。因此,制剂产品的光照试验可以在光照箱中进行,这种光照箱装有高功率紫外线发射灯并配备过滤器,能过滤320nm以下的光。