摘要
目的:探讨化学药口服固体制剂变更原料药供应商的研究流程。
方法:以苯磺酸氨氯地平片变更原料药供应商为例,考察变更后原料药的晶型、粒度分布、有机杂质、残留溶剂、致突变杂质、元素杂质;利用变更后的原料药制备3批小试样品,考察在0.01mol/L盐酸溶液、pH4.0醋酸盐缓冲液、pH5.5磷酸盐缓冲液、pH6.8磷酸盐缓冲液中的溶出曲线;进行3批工艺验证,比较原料药供应商变更前后样品中有关物质、含量均匀度、溶出度、含量等制剂关键质量指标的差异,并考察溶出曲线。
结果:供应商变更后,原料药粒度D50为9~10μm,D90为20~23μm;晶型、有机杂质基本一致;均未检出残留溶剂、致突变杂质;元素杂质含量测定结果均符合规定。3批小试样品在4种溶出介质中的溶出曲线与参比制剂和生物等效(BE)批制剂的溶出曲线均一致。3批工艺验证样品的含量、含量均匀度、溶出度分别为99.7%~99.9%、5.8%~5.9%、97%~100%,均不低于变更前的98.3%~99.5%、5.3%~5.8%、97%~99%;有关物质中,杂质D含量均为0.04%,低于变更前的0.06%~0.10%。
结论:基于苯磺酸氨氯地平片变更原料药供应商质量研究,初步建立了化学药口服固体制剂变更原料药供应商研究的基本流程。
关键词
苯磺酸氨氯地平片;化学药;口服固体制剂;药物活性成分;供应商变更;药品监管
正文
苯磺酸氨氯地平片控制血压效果较好,主要适应证为高血压、冠状动脉粥样硬化性心脏病等。由于人们不良的饮食及作息习惯,高血压患病人数呈上升趋势,且患者多为中老年人,一旦确诊需长期用药,故苯磺酸氨氯地平片等降血压类药物在国内有较大市场[1-7]。变更管理是药品生产企业质量管理的重要部分[8-12],《药品生产质量管理规范(2010年修订)》(卫生部令第79号)、美国食品和药物管理局(FDA)的《药品生产质量管理规范》(GMP)法规及欧盟GMP法规均对变更管理作了具体要求。原料药供应商变更是药品生产企业变更管理中常见的一项变更事项,原因包括原料垄断、成本考虑、原料供货及时性和可控性等,且原料药质量对制剂的安全性、有效性和质量可控性影响较大,故需对变更后原料药供应商生产的原料药进行充分研究和验证[13-17]。本研究中以苯磺酸氨氯地平片为例,探讨了化学药口服固体制剂变更原料药供应商研究的基本流程。现报道如下。
1、仪器与试药
1.1 仪器
LC-20AT型高效液相色谱仪(日本Shimadzu公司);7890B型气相色谱仪(美国Agilent公司);Bettersize 2600型激光粒度分布仪(丹东百特仪器有限公司);D8Advance型X射线衍射仪(德国Bruker公司);UDT-818A-12型溶出试验仪(禄根仪器<镇江>有限公司)。
1.2 试药
苯磺酸氨氯地平原料药(供应商A,批号分别为20AD219,20AD230,20AD241);供应商B,批号分别为C-032007004,C-032103015,C-032103016,C-032104007);苯磺酸氨氯地平片[辉瑞制药有限公司,批号为R93379(参比制剂);国药集团容生制药有限公司,批号分别为171004(生物等效批)和180401,180402,180403(供应商A),20102711,20102911,20102912(供应商B)]。
2、方法与结果
2.1 原料药
整体情况:本公司生产的苯磺酸氨氯地平片于2019年通过一致性评价,同年集中采购中标,因变更前原料药供应商为国外企业,考虑到原料药的可及性,尤其是目前全球疫情形势下境外生产原料药的风险,故原料药增加已获批的国内供应商,以保证药品安全性、有效性、质量可控性及供货稳定性。同时,依据国家药品监督管理局审评中心发布的《已上市化学药品药学变更研究技术指导原则(试行)》中第六项“变更制剂所用原料药的供应商”中所需研究验证工作及人用药品技术要求国际协调理事会(ICH)Q3中相应指导原则,选定原料药的粒度分布、晶型、有机杂质、残留溶剂、致突变杂质、元素杂质进行研究,并对制剂进行小试研究、工艺验证。供应商变更前后,合成工艺、残留溶剂、元素杂质不同,有机杂质、致突变杂质相同。详见表1。
粒度分布:溶出度是影响口服固体制剂在体内溶解、吸收的关键指标,药品溶出度与原料药的粒度联系紧密,故采用粒度分布仪对变更前后不同供应商原料药的粒径进行检测。结果见表2,其中D50和D90分别为颗粒累积分布50%和90%的粒径,供应商A和供应商B的原料药颗粒的D50和D90基本一致。
晶型:药物的晶型对制剂的溶解度和溶解速率、药物的吸收速率、药物的临床疗效及安全性等均有影响,故原料药供应商发生变更时,晶型也是需要考虑的因素之一。对苯磺酸氨氯地平原料药进行X射线衍射(XRD)分析,结果供应商变更前后原料药晶型一致。详见图1。
有机杂质:对生产工艺路线进行对比研究,确定供应商变更前后原料药中有机杂质类别;采用苯磺酸氨氯地平各国药典标准中收载的有关物质方法,对供应商变更前后的原料药进行有机杂质谱对比确认。结果变更前后有机杂质一致(表3),并按药典标准对各杂质进行了确认(图2和图3)。其中,供应商变更前,3批(批号分别为20AD219,20AD230,20AD241)原料药有关物质I于254nm和365nm波长处检测均符合规定。
残留溶剂:根据供应商变更后原料药的工艺流程,确定可能引入的残留溶剂,并对原料药中残留溶剂及供应商B生产的多批次原料药进行研究。结果供应商变更后,4批(批号分别为C-032007004,C-032103015,C-032103016,C-032104007)原料药中均未检出残留溶剂乙醇、乙酸乙酯、苯、甲苯、N,N-二甲基甲酰胺、冰醋酸,表明其生产过程中残留溶剂控制较好。
致突变杂质:根据ICH M7指南及供应商原料药的工艺流程,变更供应商后对致突变杂质进行研究。依据ICH M7(R1)指南,按毒理学的关注阈值(TTC)的可摄入量为1.5g/d进行计算,苯磺酸氨氯地平片每日最大剂量为10mg,苯磺酸甲酯、苯磺酸乙酯、3-氨基巴豆酸甲酯的限度分别为75×10-6,75×10-6,50×10-6。结果供应商变更后,4批(批号分别为C-032007004,C-032103015,C-032103016,C-032104007)原料药中均未检出苯磺酸甲酯、苯磺酸乙酯、3-氨基巴豆酸甲酯,表明原料药中致突变杂质控制较好。
元素杂质:根据ICH Q3D指导原则及原料药合成路线,对供应商B提供的原料药进行元素杂质研究,因供应商B在原料药生产过程中未使用金属催化剂,故仅检测1类及2A类元素杂质。按ICH Q3D(R)元素杂质指导原则,钒(V)、钴(Co)、镍(Ni)、砷(As)、镉(Cd)、汞(Hg)、铅(Pb)的限度分别为不超过10000×10-6,5000×10-6,20000×10-6,1500×10-6,500×10-6,3000×10-6,500×10-6。苯磺酸氨氯地平片每日最大剂量为10mg,计算原料药中上述元素杂质的限度。结果供应商变更后,原料药中元素杂质含量均符合标准。详见表4。
2.2 制剂
小试研究:采用供应商B提供的原料药进行小试研究,按原处方工艺制备3批(批号分别为20032401,20032701,20032702)小试样品。同时,依据《已上市化学药品药学变更研究技术指导原则(试行)》中第六项“变更制剂所用原料药的供应商”中所需研究验证工作中“变更前后样品的溶出曲线、关键理化性质应保持一致”等规定进行体外释放研究。参考原国家食品药品监督管理总局发布的《普通口服固体制剂溶出度试验技术指导原则》(2015年第3号)、《普通口服固体参比制剂选择和确定指导原则》(2016年第61号),比较3批小试样品、参比制剂(批号为R93379)及BE批(批号为171004)样品在0.01mol/L盐酸溶液、pH4.0醋酸盐缓冲液、pH5.5磷酸盐缓冲液、pH6.8磷酸盐缓冲液中的溶出曲线。结果供应商变更后,小试样品在4种溶出介质中的溶出与参比制剂及BE批制剂一致,表明原料药供应商变更后,生产的小试样品检测结果均符合质量标准。详见图4。
A. 0. 01 mol / L hydrochloric acid solution B. pH 4. 0 acetate buffer C. pH 5. 5 phosphate buffer D. pH 6. 8 phosphate buffer
Fig. 4 Dissolution profiles of small test sample,reference preparation,and BE batch preparation in four dissolution media
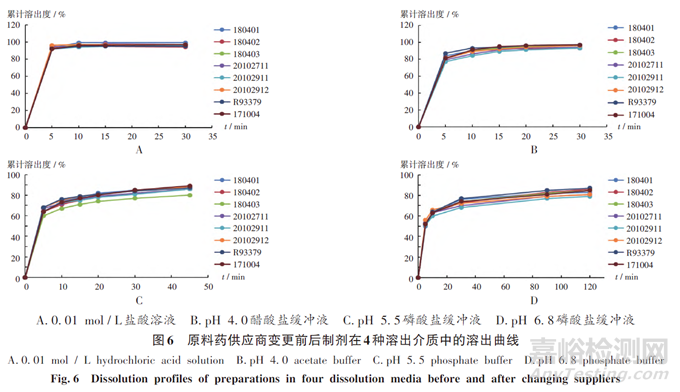
工艺验证:原料药供应商变更前后,制剂的生产信息(生产商、批量、批处方、生产工艺、工艺流程图、生产设备、关键工艺步骤、关键工艺参数、中间体控制等)一致,考察变更前后制剂的关键理化性质和溶出曲线。参考国家药品监督管理局标准YBH06412019,考察有关物质、含量均匀度、溶出度、含量。其中,有关物质杂质D、其他单个杂质、其他总杂质的含量分别应不超过0.2%,0.2%,0.5%;含量均匀度应不超过15.0%;溶出度应不低于标示量的80%;按氨氯地平(C20H25ClN2O5)计算,含量应为标示量的95.0%~105.0%。结果见图5和图6,表明原料药供应商变更后,生产的制剂质量标准不低于变更前。
3、讨论
由粒度及晶型XRD图谱可知,供应商变更前后的原料药关键理化性质一致;对变更前后原料药的合成路线及杂质谱进行对比分析可知,变更前后杂质谱一致,且采用各国药典标准中收载的有关物质检测方法进行检测,结果显示,变更后的杂质检出个数及杂质检出量均低于变更前;对供应商B提供的原料药残留溶剂、致突变杂质、元素杂质进行研究可知,因合成工艺不同,其残留溶剂及元素杂质与供应商A提供的原料药有所不同,但可通过相应研究进行控制,致突变杂质均未检出,初步表明变更后供应商B提供的原料药优于或等同于变更前的原料药。采用供应商B提供的原料药进行小试研究,小试样品各项检测结果均符合质量标准规定,在不同介质中的溶出曲线与BE批及参比制剂均基本一致,表明原料药供应商可替换。同时,进行3批工艺验证,并进行质量检测及溶出曲线研究,结果显示,原料药供应商变更前后,制剂的性状、含量、含量均匀度、溶出度、有关物质均一致,在4种溶出介质(0.01mol/L盐酸溶液、pH4.0醋酸盐缓冲液、pH5.5磷酸盐缓冲液、pH6.8磷酸盐缓冲液)中的溶出曲线均一致,表明用供应商B提供的原料药生产的苯磺酸氨氯地平片与变更前质量一致。
以苯磺酸氨氯地平片变更原料药供应商为例,初步建立了固体口服制剂改变制剂药物活性成分供应商的基本流程。第一步,进行原料药质量研究(如粒度、晶型、有机杂质、残留溶剂、致突变杂质、元素杂质),并保证原料药质量基本一致;第二步,进行制剂小试研究,并进行体外释放试验,保证变更前后小试样品与参比制剂及BE批相似;第三步,制备3批样品进行制剂工艺验证,按相关质量标准进行检测,保证变更后制剂质量不低于变更前,并通过原料药质量研究及成品检测研究过程证明流程的科学性与合理性。
参考文献:详见《中国药业》2023年11月5日 第32卷第21期