杂质在药物研发或生产过程中几乎是不可避免的,但杂质并不能为患者提供获益,反而会引入安全性风险,因此需要对其限度进行严格控制。杂质种类很多,根据来源分类有工艺相关杂质、降解产生的杂质、起始物料或试剂中混入的杂质等等。按照理化性质可分为有机杂质、无机杂质和残留溶剂。有机杂质可能会在新的原料药的生产过程和/或储存期间产生,结构可能已鉴定,也可能未鉴定。无机杂质通常来源于生产过程,结构已知。溶剂则指新原料药合成过程中用到的有机或无机液体,毒性已知。其中,基因毒性相关杂质的评估和控制可以参照ICH M7,残留溶剂的要求可以参照ICH Q3C(有些残留溶剂是不被允许使用的,建议读者详细阅读指南),元素杂质每日允许暴露量等要求可以参考ICH Q3D。
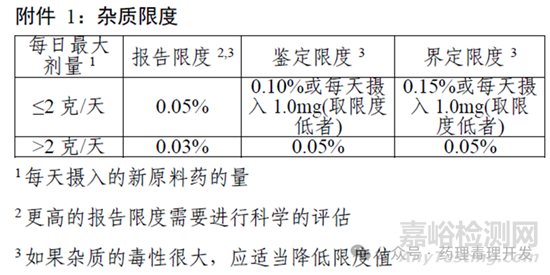
ICH Q3A和Q3B对非基因毒性杂质在原料药(DS)和制剂(DP)中的限度进行了规定。其中很重要的一个概念是界定限,低于该限度则可视为不会产生额外毒性。比如,如果一个药物每日最大剂量≤2g,DS中非基因毒性杂质的界定限度为1mg/天或0.15%,哪个数值小则以哪个为准。比如,某药物临床每日最大剂量为1.5g/天,按照0.15%界定限度计算,则杂质摄入的绝对量为2.25mg,超过1mg,则取限度更低的1mg设定限度,1mg÷1.5g×100%=0.07%。如果超出这一限度,且无更多数据可判定该杂质的安全性,则需要开展非临床研究确定其潜在风险。ICH Q3A中关于杂质限度的示例如下:
ICH Q3A、Q3B的要求更多适用于晚期临床阶段的产品,对于早期临床阶段药物的非基因毒性杂质要求尚无指南出台。对于没有指南的情况,企业通常也会遵循一定的路径去制定一个可接受安全性限度。通常采用的是如下几种方案:
1)毒理批和早期临床研究采用同一批次的产品,这种方法看似简单可行,并解决了部分问题,比如减少了批次间差异引入的风险。但不太实用,有可能会导致临床批样品很快不够,还是要制备新批次样品,依然存在新批次可能出现新杂质的风险。
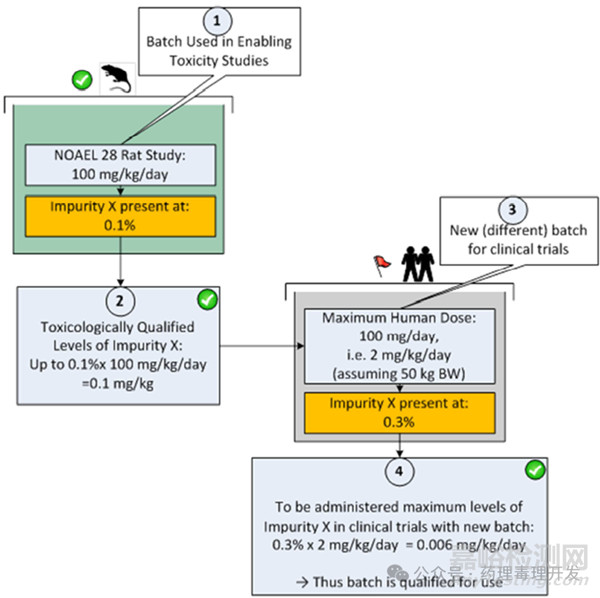
2)基于非临床研究毒理学数据和预期临床每日最大用量进行计算,如下图所示。这种方法比较常用一些。先根据非临床动物毒理研究的NOAEL及杂质含量,确定杂质的NOAEL剂量。再计算临床新批次的杂质含量在临床最大拟用剂量下的杂质剂量。如果临床新批次样品计算出的剂量小于非临床杂质NOAEL水平,那下图中的0.3%的新批次限度也是可以接受的。如果像肿瘤产品无法确定NOAEL,可根据具体情况选择LOAEL、STD10或HNSTD来进行计算。
同理,也可以通过以上思路确定非基因毒性杂质限度,动物毒理NOAEL(mg/kg)×杂质含量(%)×人体体重(如50kg)÷临床最大拟用剂量(mg)×100%=新的限度。
当然,这里面的算法可能很多朋友会提出挑战,因为未进行种属间的基于体表面积(BSA)的剂量换算。看一下其它口径信息的一些观点。
CDE黄晓龙老师2007年发表的《对创新药研发中杂质限度确定的几点思考》一文中有个案例,某原料药杂质A的含量为0.12%,犬6个月长毒剂量为5、10、40mg/kg/d。本品的临床剂量为160mg/d,患者体重以60kg计,人体给药剂量为160/60=2.67mg/kg。与犬的给药剂量比较,则该杂质的安全限度应为(40/2.67)*0.12%=1.8%。由于文中未给出详细说明,暂不讨论选择40mg/kg的原因。更关心的点是,此处也未进行种属间基于BSA的剂量折算。
CDE何伍、王海学老师2009年发表的《浅谈药物杂质限度的制订方法》一文中分享过类似案例。杂质A毒理批限度0.34%,临床批限度调整为0.5%,远高于ICH Q3A规定的限度。计算依据如下:该产品大鼠长毒NOAEL剂量为26.5mg/kg/d,对应的杂质A的剂量水平为26.5*0.34%=0.09mg/kg/d,根据动物类比法转换为人体的剂量水平为0.0146mg/kg/d。人体按照60kg计,0.0146*60=0.88mg/d。人体最大拟用量为10mg/人,按照临床批限度0.5%计算,杂质A人体每天用量为0.05mg/天,远小于动物试验转化获得的0.88 mg/d。与前述文献不同的是,本文是进行了基于BSA的种属间剂量换算。
2023年4月26日,CDE姚方耀老师分享了《新药临床试验申请前药学沟通交流技术要求及案例分析》这一主题,在杂质研究部分有提到对于实测值较高的杂质,需要基于药理毒理研究数据提供计算方式和依据。
同时,姚老师还给出了一个具体案例,采用非临床研究中两个种属的NOAEL剂量,毒理批杂质的水平,再结合临床方案中最大给药剂量,得出基于动物的人体最大可接受限度。但由于案例中未给出具体NOAEL剂量,不确定种属间剂量换算的方式。
几种算法最大的区别就是杂质的剂量是否需要进行种属间的剂量换算。不换算的杂质限度会更高,根据采用的NOAEL剂量来自大鼠还是比格犬,亦或其它,相差倍数不等。
关于这点,来自BMS、Merck、Novartis、AbbVie、Pfizer、GSK、AZ和Gilead团队组成的IQ DruSafe Impurities Working Group进行过一次调研,结果于2021年公布,如下图所示,44%(10/23)的企业采用mg/kg,39%(9/23)的企业选用的BSA。未采用BSA进行换算的企业中,59%并未受到监管机构的挑战,41%则被要求说明不选择BSA的原因。当然,在本文献最后,作者总结到,杂质限可根据mg/kg去界定,并认为从安全性角度看,基于像BSA的转换,似乎没有必要。
3)非临床研究中作为母体代谢产物存在的杂质限度确定。《化学药物杂质研究技术指导原则》中有相关描述“如某杂质同时也是该药物在动物或人体中的主要代谢产物,则对该杂质可不考虑其安全性,但需制订合理的限度”。ICH Q3A(R2)和Q3B(R2)中也提到,“对于一个通过充分的安全性研究和临床研究的新原料药,其中任何一个杂质的水平应被认为是已经通过合理界定的。对于是动物和/或人体中的重要代谢物的杂质,通常也视为已通过界定。杂质的界定限量(水平)如果高于药物实际所含的杂质量,则同样可以根据对已完成的安全性研究中使用药物中的实际杂质量来判断其合理性。”当然,对于药物代谢产物的评价也要具体情况具体分析,《药物代谢产物安全性试验技术指导原则》中也提出,“如果某种代谢产物仅在人体中出现而在受试动物种属中不存在,或者某种代谢产物在人体的暴露比例水平高于采用母体药物进行标准毒理学试验的动物种属中的暴露比例水平时,代谢产物的安全性就值得关注,应考虑进行代谢产物的非临床安全性评价。”大家可去查阅参考。
4)通过药物类别和临床使用经历,根据对安全性担忧的程度,合理调整杂质限度。如抗肿瘤药物,可参照ICH S9中关于杂质限度的要求,是有可能接受超出ICH Q3A和Q3B确定的杂质限度的。且可以根据具体情况选择LOAEL、STD10或HNSTD来进行计算。
但是,以上4种方案并不能覆盖药物开发过程中遇到的所有场景,比如说临床批样品出现了毒理批未发现的新的杂质,就无法通过临床前动物试验数据进行推导。最佳的方案还是像ICH Q3A和Q3B那样,给出一个具体的控制限度。那早期临床阶段的样品杂质限度可以适当放宽到什么程度才比较合理呢?
2017年,来自GSK、Pfizer、AZ、Janssen团队Harvey等人联合发表了一篇《Management of organic impurities in small molecule medicinal products: deriving safe limits for use in early development》,对早期临床阶段的化药杂质限度进行了讨论。大部分药物的毒性是剂量和给药周期依赖的,因此与晚期阶段的界定限相比,早期临床阶段的可以更高。当然,如果药物整个开发生命周期均按照ICH Q3A中的杂质限度进行控制,由杂质引发的安全性风险默认为会比较低。
ICH并未给出每日最大剂量≤2g/天时,鉴定限度定为0.1%或1mg的依据。不过,按照人体50kg计,1mg/天对应的剂量为0.02mg/kg/天。Munro数据库包含613个化合物,95%化合物的NOELs>0.22mg/kg/天,比ICH制定的限度高11倍。eTOX数据库更是显示100%的化合物NOELs≥0.02mg/kg/天。当然,还有很多团队检索调研过已公开化合物的NOELs水平,结论都是一致的,ICH制定的杂质限度是非常安全且保守的。
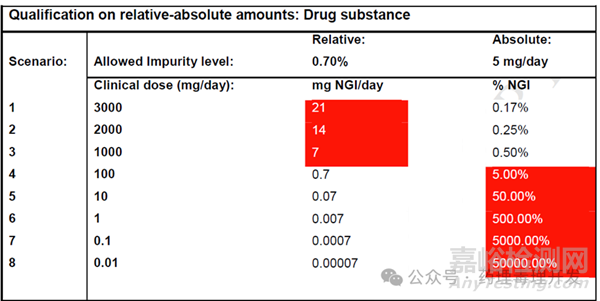
ICH的杂质限度针对的是长期用药,甚至说是终生服药的情况(safe dose of lifetime exposure),那么对于早期给药周期<6个月的产品,这一阈值可以放宽到什么程度呢。有一种算法称之为modification of Haber’s Law。根据这一计算方式,按照1mg/天的终生暴露水平计算,<6个月的暴露水平对应的杂质限度可提高到5mg/天,计算过程如下图所示。同样的,界定限度也从0.15%升高只0.7%。
具体以5mg还是0.7%为准,与ICH原则一致,以数值小为准,举例如下图所示,100mg及以下每天用量,以0.7%为准,1000mg及以上用量则以5mg/天计算限度。该文章同样拓宽制剂限度至5mg或2%,原理类似,篇幅所限,不再展开。
工业界这四个MNC企业把限度制定好了,也拓宽了ICH规定的的边界,但监管机构认可不认可呢?会不会被认为在故意降低质量标准,不被监管机构接受呢?
2024年,AZ和Ipsen团队发表了一篇《Non-mutagenic impurities-Recent industry experience of using dose durational limits in drug development》,总结了来自European Federation of Pharmaceutical Industries and Associations (EFPIA)的六个成员企业关于非基因毒性杂质超出ICH Q3A和Q3B规定的限度后,与监管机构的沟通案例。下图中的5个案例,均处于早期临床研究阶段,3个肿瘤适应症,2个非肿瘤适应症,要么超限,要么出现了新的杂质,监管机构要求对不符合ICH Q3A和Q3B的要求进行解释。其中4个采用前文所述的Harvey等人发布的限度进行解释,被监管机构接受。
当然,无论采用非临床数据进行的推算,还是采用Harvey等人的更宽泛的限度建议,这都是对早期临床样品的包容度。随着研发的推进,杂质限度要求还是要朝着ICH规定的方向靠拢和努力。
最后
化学药物彼此之间千差万别,ICH规定的限度对于绝大多数杂质来讲,都是相对安全的,能适应绝大多数场景,并能更大程度平衡获益/风险,但是更适合晚期临床阶段的产品。早期临床工艺尚未完全确定,杂质研究还处于逐渐发现和优化的阶段,杂质限度往往做不到完全符合ICH相关规定。基于这个背景,我们总结了国内外监管机构和工业界对这类情况的处理和建议。当然,由于缺少官方特定指南,大家对超出ICH Q3A&Q3B要求的非基因毒性杂质限度的处理方式难免不一。核心的思路大概有如下两种:
1)基于非临床毒理研究数据和毒理批次杂质含量,结合临床拟用最大剂量进行计算。但这一思路需要注意一些细节,比如是否需要基于BSA进行种属间的剂量换算,关于这点无论是国外(39%用,44%不用)还是国内(黄晓龙老师和何伍老师的两篇文章之间的案例),是有些分歧的。比如是否是原型药物代谢产物,代谢产物需要结合非临床代谢和毒理数据评估。比如是否是肿瘤药物,肿瘤药物很少获得NOAEL,可以考虑用LOAEL、STD10或HNSTD替代。
2)Harvey等人制定的绝对限度,比ICH限度宽5倍左右,可以作为早期临床阶段药物非基因毒性杂质限度的参考,已经有被监管机构接受的多个案例。
与监管机构沟通时,可以考虑多路径,多维度对非基因毒性杂质限度进行计算,提供充分且合理的证据。
当然,有些非基因毒性杂质的限度不仅不能放宽,甚至需要考虑缩窄。为什么说ICH Q3A&Q3B适用于绝大多数杂质,而不是所有杂质,因为总有一些特殊情况。比如,如果基于已有数据或同类数据,一些杂质被认为具有异乎寻常的活性或毒性、与免疫调节相关或临床担忧等,ICH的限度也不安全,需要根据实际情况合理说明。
最后提醒一点,产品的杂质限度往往用于生产检验,但对于降解杂质的限度,应该是货架期终点的最高限度,因而需要被限制的是整个有效期内针对典型条件都不应超过这个限度,而不仅仅是生产出来做质量检验时不超过该限度。因此,需要从整个产品的生命周期思考杂质限度的问题。