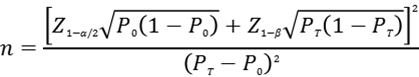结直肠癌筛查用体外诊断试剂临床评价注册审查指导原则
(征求意见稿)
本指导原则旨在指导注册申请人对结直肠癌筛查试剂注册申报资料的准备及撰写,同时也为技术审评部门对注册申报资料的技术审评提供参考。
本指导原则是针对结直肠癌筛查试剂注册审查的一般要求,申请人应依据产品的具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。
本指导原则是供注册申请人和技术审评人员使用的指导性文件,但不包括审评审批所涉及的行政事项,亦不作为法规强制执行,应在遵循相关法规的前提下使用本审评要点。如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
本审评要点是在现行法规和标准体系以及当前认知水平下制定,随着法规和标准的不断完善,以及科学技术的不断发展,相关内容也将适时进行调整。
一、适用范围
本指导原则适用于对来源于粪便样本中的相关标志物进行体外定性检测,用于结直肠癌筛查的体外诊断试剂。阳性结果提示存在结直肠癌或进展期腺瘤的风险较高,应进行诊断性结肠镜检查。产品检测结果不能替代肠镜。对于其他样本类型,可能部分要求不完全适用或本文所述内容不够全面,申请人可以参照本指导原则,根据产品特性对适用部分进行评价或补充其他的评价资料进行相应验证。
二、临床试验注册审查要点
开展结直肠癌筛查试剂临床试验应符合《体外诊断试剂注册与备案管理办法》《医疗器械临床试验质量管理规范》以及《体外诊断试剂临床试验技术指导原则》的要求,如相关法规、规章、规范性文件有更新,临床试验应符合更新后的要求。
申请人应根据产品特点及预期用途,综合不同地区发病情况等因素选择具有代表性的临床试验机构开展临床试验,保证入组人群对筛查目标人群总体具有良好的代表性。应选择不少于3家(含3家)符合法规要求的临床试验机构开展多中心临床试验。
参与临床试验的人员经培训后应熟悉相关检测技术的原理、适用范围、操作方法等,并能够对检测结果进行正确判读。在整个试验中,试验体外诊断试剂和对比试剂/方法都应处于有效的质量控制下,最大限度保证试验数据的准确性及可重复性。
临床试验应包括:对目标人群的筛查性能评价;对结直肠癌诊断的灵敏度和特异度评价;被测标志物的检测性能评价等。
(一)对目标人群的筛查性能评价
1.受试者入组
筛查临床试验在预期适用的目标人群中按照临床试验方案设定的入组标准前瞻性、顺序入组受试者。
应明确人群的判定依据。根据《中国结直肠癌筛查与早诊早治指南(2020,北京)》,结直肠癌高风险人群包括有结直肠癌病史、腺瘤性息肉病史、炎症性肠病史、有提高结直肠癌罹患风险的已知疾病史或家族遗传疾病史(如林奇综合征或家族性腺瘤样息肉病)、一级亲属结直肠癌病史以及粪便隐血试验阳性的人群。一般风险人群为40-74岁之间的无结直肠癌高风险因素的人群。有关高风险人群和一般风险人群的定义建议按照现行有效的筛查指南执行。
受试者有无高风险因素等临床背景可采用问卷方式获得,并在临床试验数据表中针对每一项风险因素单独设一列,填写问卷结果。
2.试验方法
采用试验体外诊断试剂与结直肠癌和进展期腺瘤临床诊断参考标准,即结肠镜结合病理检查结果进行比较研究,评价试验体外诊断试剂在适用人群中筛查结直肠癌和进展期腺瘤的灵敏度、特异度和阳性/阴性预测值等指标。
如临床已有相同适用人群的筛查方法,应同时采用已有方法进行筛查,评价现有方法的临床性能,并与试验体外诊断试剂进行比较。
受试者入组后采用试验体外诊断试剂和现行指南推荐的筛查方法分别进行检测/检查,之后进行结肠镜检查,必要时进行病理检查。试验体外诊断试剂与已有筛查方法的样本采集应在结肠镜检查之前完成,与结肠镜检查的时间间隔建议不超过12周。结肠镜检查质量应符合高质量检查标准(良好的肠道准备率应>85%;盲肠插镜率>95%;退镜时间应至少保证6min)。
3.样本量
应根据预期灵敏度水平,采用单组目标值法样本量公式估算最低结直肠癌样本例数。
公式中,n为阳性样本量;Z1-α/2、Z1-β为显著性水平和把握度的标准正态分布的分位数,P0为评价指标的临床可接受标准,PT为试验体外诊断试剂评价指标预期值。
基于现有认知,结直肠癌灵敏度P0建议不低于85%。
按照估算的最低结直肠癌样本例数和预期适用人群中结直肠癌患病率估算总样本量,同时考虑可能的受试者脱落比例,确定需要入组的受试者最低总样本量,脱落率建议不高于20%。
4.评价指标和统计学分析
首先应对入组人群基线信息进行总结分析,包括人口学信息,如年龄、性别、地域(农村,城市),以及风险因素分布等。
其次建议采用流程图的形式说明入组人群、脱落人群、纳入统计人群的情况,并对所有脱落病例进行分析,解释脱落原因和脱落率可接受的理由。
最后对筛查试验的各项临床性能指标进行分析,一般包括临床灵敏度、临床特异度、阳性预期值、阴性预期值、相对风险值等。其中临床灵敏度应针对结直肠癌和进展期腺瘤病例组分别评价,临床特异度应针对非结直肠癌且非进展期腺瘤受试者进行评价。应针对各项评价指标进行点估计值及95%置信区间计算。同时计算入组人群中结直肠癌患者占比,包括各分期患者占比,应与同时期流行病学调查结果相符。
临床试验结果汇总和各项指标计算方法参见表1。
表1临床试验检测结果总结
|
试验体外诊断试剂检测结果
|
结肠镜结合组织病理学检查结果
|
|
|
结直肠癌
|
进展期腺瘤
|
中风险腺瘤
|
低风险腺瘤
|
无发现
|
合计
|
|
阳性
|
a
|
b
|
c
|
d
|
e
|
a+b+c+d+e
|
|
阴性
|
f
|
g
|
h
|
i
|
j
|
f+g+h+i+j
|
|
合计
|
a+f
|
b+g
|
c+h
|
d+i
|
e+j
|
N
|
注:1.结直肠癌:I-IV期结肠直肠癌。
2.进展期腺瘤:高级别瘤变或10个以上腺瘤;管绒毛腺瘤;≥10毫米的管状腺瘤;≥10毫米的传统的锯齿状腺瘤。
3.中风险腺瘤:≥10毫米的增生性息肉或SSL;<10毫米的5-9个腺瘤(TA + SSL);<10毫米的3-4个腺瘤(TA + SSL)。
4.低风险腺瘤:1-2个5-9毫米的腺瘤(TA + SSL);1-2个<5毫米的腺瘤(TA + SSL)。
5.无发现:<10毫米的增生性息肉等阴性病变或结肠镜检查无病变。
结直肠癌临床灵敏度=a/(a+f)×100%
进展期腺瘤临床灵敏度=b/(b+g)×100%
非进展期腺瘤临床灵敏度=(c+d)/(c+d+h+i)×100%
临床特异度=(h+i+j)/(c+d+e+h+i+j)×100%
阳性预测值=(a+b)/(a+b+c+d+e)×100%
阴性预测值=(h+i+j)/(f+g+h+i+j)×100%
相对风险=阳性预测值/(1-阴性预测值)
如适用人群为满足一定年龄、性别要求的全人群,建议针对其中的一般风险人群和高风险人群进行分层分析,确认不同人群的筛查性能均满足临床要求。
产品如包括不同的标志物,应包括不同标志物的筛查灵敏度分析,确认不同标志物联合检测的意义(如适用)。如果试验体外诊断试剂不适用与此项评价,应有充分的理由。
同步开展的已有筛查方法检测/检查,应参照上述方法同步评价各项临床性能指标。
最后应将试验体外诊断试剂在适用人群中筛查结直肠癌和进展期腺瘤的各项临床性能指标与已有筛查方法各项临床性能指标进行比较,并分析申报产品临床性能指标能否满足临床需求。
(二)结直肠癌及癌前病变诊断的灵敏度和特异度评价
1.受试者入组
为更加充分的评价试验体外诊断试剂的灵敏度和特异度,还应对肿瘤组和非肿瘤组的重要亚组分别进行分层入组,并与相关结直肠癌的临床参考标准进行对比,从而使临床性能评价更加全面地覆盖目标人群中的各种特征,包括不同分期的肿瘤病例,和不同干扰因素的非肿瘤病例等。此部分试验可将(一)筛查性能评价中顺序入组的所有样本纳入,如某些亚组样本量不足可进一步富集入组。
1.1肿瘤疾病组
肿瘤疾病组人群应包括结直肠癌不同分期(Ⅰ、Ⅱ、Ⅲ、Ⅳ期)、结直肠进展期腺瘤。应特别关注进展期腺瘤及早期癌症组的临床灵敏度是否满足要求。
1.2非肿瘤疾病组
为对产品临床特异度进行充分评价,应纳入消化道良性疾病患者(肠息肉、肠腺瘤、肠炎等)和其他消化道肿瘤患者包括胃癌、肝癌、食管癌、胆管癌、胰腺癌等。非结直肠癌肿瘤疾病患者的诊断依据相关疾病诊疗规范执行。
2.试验方法
采用试验体外诊断试剂与结直肠癌和进展期腺瘤临床诊断参考标准进行比较研究,评价试验体外诊断试剂针对结直肠癌各分期、进展期腺瘤疾病组的临床灵敏度,以及消化道良性疾病、其他肿瘤亚组的临床特异度等指标。
3.样本量
可采用单组目标值法公式估算最低样本量,应分别估算结直肠癌患者例数、进展期腺瘤患者例数、消化道良性疾病患者例数和其他癌症患者例数等)以及肠道无占位性病变病例数。各组目标值设定建议参考表2中相关指标的最低可接受标准。
根据已有文献数据,经统计学估算,建议结直肠癌病例不少于300例,其中各分期病例建议I期不少于80例,其他各期分别不少于50例;进展期腺瘤不少于200例;肠道良性疾病患者中,肠息肉患者不少于200例;其他各种癌症病例建议分别不少于30例,无肠道异常病例不少于230例。
4.临床评价指标和统计学分析
对临床试验结果进行总结。计算灵敏度、特异度并计算95%置信区间;同时应针对结直肠癌不同分期、以及进展期腺瘤患者分别计算灵敏度;针对不同类型肠道良性疾病患者、其他不同癌种病例以及肠道无占位性病变患者分别分析特异度。
依据临床需求和已报道研究数据,部分关键亚组临床性能评价指标推荐的最低可接受标准(95%置信区间下限)参下表:
表2部分亚组临床性能评价指标推荐最低可接受标准
|
亚组
|
评价指标
|
预期可接受标准
|
|
CRC
|
临床灵敏度
|
85%
|
|
进展期腺瘤
|
临床灵敏度
|
50%
|
|
肠道良性疾病
|
临床特异度
|
80%
|
|
其他恶性肿瘤
|
临床特异度
|
80%
|
|
无肠道异常
|
临床特异度
|
90%
|
对于检测结果与临床诊断结论不一致的样本,应结合患者疾病背景信息、其他临床实验室检验结果等对差异原因进行合理分析。
(三)被测标志物检测性能评价
1.试验方法
根据被测标志物的特征,应采用试验体外诊断试剂与已上市同类产品或实验室参考方法进行对比试验,评价试验体外诊断试剂针对被测标志物的检测性能。如被测标志物为DNA甲基化或者基因突变,可以采用测序方法作为实验室参考方法,进行对比试验,如选择同类产品,应确认具有良好的可比性。实验室参考方法应进行充分的性能验证,包括最低检出限、准确性、精密度等,确认与试验体外诊断试剂具有良好的可比性。并提供详细的方法建立和性能验证资料。如测序试验委托第三方实验室或检测机构完成,应提供临床试验机构委托该试验的委托协议,并确认第三方检测机构有相应的检测资质。
当试验体外诊断试剂包含多项被测物时,原则上应针对所有标志物分别进行检测性能评价。如不适用,应提供充分的理由和依据,并采用合理的方法进行检测性能评价。
2.样本量
应针对所有标志物(如适用)分别估算阳性样本量和阴性样本量,建议采用单组目标值法公式,目标值(P0)设定应有合理依据,基因突变或甲基化等核酸标志物检测目标值建议不低于90%。
3.评价指标和统计学分析
对于试验体外诊断试剂与对比试剂/方法的一致性评价,可采用四格表的形式总结两种试剂/方法的检测结果,计算阳性符合率、阴性符合率、总符合率及其95%置信区间。
对于不一致样本,应结合患者便隐血结果、结肠镜和病理诊断结果进行原因分析。
(四)筛查频率
申请人应基于已有研究成果和文献数据针对筛查频率给出合理建议,同时应于产品上市后持续开展研究,分析不同筛查频率下的临床性能,并与已有筛查方法进行比较,需要时进一步优化筛查频率。
(五)其他评价
在对申报产品各项临床性能进行充分评价的基础上,建议申请人在临床试验过程中对试验体外诊断试剂预期用于目标人群的成本效益比以及受试者接受度等进行分析,结合产品临床性能指标综合评价产品对筛查人群可能带来的风险和获益,并与已有筛查方法的风险和获益进行比较,评价申报产品的优势。
三、参考文献
[1]体外诊断试剂注册与备案管理办法[Z].
[2]医疗器械临床试验质量管理规范[Z].
[3]体外诊断试剂临床试验技术指导原则[Z].
[4]中国结直肠癌筛查与早诊早治指南 (2020,北京)[Z].
[5]中国结直肠癌诊疗规范(2023版)国家卫生健康委员会[Z].
[6]U.S. Preventive Services Task Force Recommendation Statement. Colorectal Cancer: Screening .[Z].
[7]Weiderpass E, Stewart BW. World Cancer Report: Cancer Research for Cancer PreventionWild CP .[Z]..
[8]詹思延.流行病学[M].8版.北京:人民卫生出版社, 2017: 120-139.[Z].