摘 要 Abstract
药品开发,必须符合相关法规要求,需要以监管机构制定的行业指南为指针,参考监管机构制定的相关用例。新药获批必须基于临床试验生成的证据。随着生物制药行业的不断发展,临床试验复杂程度增加。单纯依靠增加投入,难以改善临床试验效率,解决管线产出率低、成本高企的问题。本文通过解析FDA 讨论文件、EMA 观点文件中涉及的临床试验中人工智能/ 机器学习(AI/ML)运用内容,以及介绍利益攸关方对FDA 讨论文件的反馈意见,初步探讨AI/ML 在临床试验中的运用以及监管挑战。
Drug development must comply with relevant regulatory requirements, guided by industry guidelines developed by regulatory agencies, with reference to relevant use cases. The approval of new drugs must be based on evidence generated from clinical trials. With the continuous development of biopharmaceutical industry, the complexity of clinical trials has increased. Merely increasing investment is insufficient for improving clinical trial efficiency and addressing issues of low pipeline productivity and soaring costs. This paper explores the application and regulatory challenges of artificial intelligence/ machine learning (AI/ML) in clinical trials by analyzing content from FDA discussion papers, EMA reflection papers, and stakeholders’ comments.
关键词 Key words
人工智能;机器学习;临床试验;方案设计;试验效率;管线产出率;透明度
artificial intelligence; machine learning; clinical trials; protocol design; clinical trial efficiency; pipeline yield; transparency
近年来新药研发投入不断增长,但新药临床试验损耗率并未相应下降,研发管线产出率没有显著增长,导致新药研发风险加大,费用高企。根据TrialTrove数据估算的临床试验成功率和获批成功率显示,截至2022 年第一季度,进入临床试验的在研药品中,从Ⅰ期至获批的成功率仅为10.3%, 其中肿瘤药仅为7.2%,罕见病用药为11.0%( 图1 和图2)。如何改善临床试验效率,成为业界关注的焦点之一。
传统的线性、序贯临床试验,被公认为是确保新药有效性和安全性的方法。一直被视为金标准的随机对照试验,主要是为检测面向大众市场的在研药品设计的。患有同一种疾病的患者,不同个体或不同部位可出现不同的临床表现或病理特征,表现出临床异质性。癌细胞、免疫细胞也往往具有异质性特征。随着个体化用药时代的到来,制药行业开发的很多新药所针对的适应症患者群体越来越小,传统的基于群体临床试验的监管审评方式,不完全适用于个体化用药与罕见病用药。随机对照试验缺乏开发复杂新疗法所需的分析能力、灵活性和速度。患者选择、招募和保留困难,再加上难以有效管理和监测参与临床试验的患者,导致进入临床试验后的研发管线损耗率居高不下,研发管线从进入Ⅰ期临床试验到获得监管机构批准的成功率极低,推高研发成本。
人工智能(artificial intelligence,AI)医疗应用增长的驱动力包括:①大型数据集(例如管理数据、电子病历、登记等)。②多样化和多模态数据集(例如分布式哈希表、基因组、检验所、医学成像等)。③数据标准改进(例如ICD-10、LOINC、NDC、UMLS、FHIR/HL7、OHDSI 等)[1]。④数据互操作性和医疗数据交换改进。⑤算力增强。⑥数据隐私权保护方法取得进展。⑦相关方法取得突破(例如深度神经网络、强化学习、生成对抗网络、变异自动编码器等,以及因果推理方法,例如结构因果模型和因果贝叶斯网络等领域的突破)。
利用预测性AI 模型和分析工具挖掘真实世界数据,可以加快对疾病的了解,确定合适的患者和关键研究人员,为试验场所提供依据,支持新型临床研究设计。AI 算法与有效的数字底层架构结合,可以清理、汇总、编码、存储和管理源源不断的临床试验数据。减少数据采集中人为差错的影响,促进与其他数据库无缝集成。人工智能/ 机器学习(AI/ML)可用于剂量优化,临床试验设计,患者招募与选择,患者黏度,患者保持,临床试验场所选择,数据采集、管理、分析,临床终点评价[2], 战略性合作伙伴选择,组织内AI/ML 知识培训[3]。AI 赋能技术在临床试验各个环节中的运用详见图3。
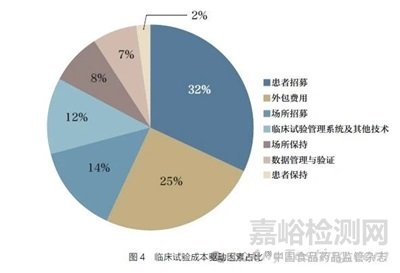
尽管随机对照试验仍被认为是验证新活性实体在大量人群中的有效性和安全性的金标准。但在可互操作数据、开放和安全平台、消费者驱动的医疗保健以及从医疗保健到健康的根本性转变的推动下,生命科学和医疗保健行业正处于大规模颠覆的边缘。临床试验将需要更多更具针对性的方法。采用AI 支持的数字医疗技术和患者支持平台,有望使临床试验发生改变,在整个研究期间和研究终止后吸引和留住患者,提高临床试验效率。AI、计算机模拟和个体化医疗的发展将导致计算机模拟试验兴起,在药品开发或监管评价过程中使用先进的计算机建模和模拟。新冠疫情大流行加速了对虚拟临床试验和分散式临床试验的需求,这些试验将利用创新数字技术减轻患者的经济和时间负担。虚拟试验可在患者日常生活环境中实时、更快地招募更具代表性的群体,远程监测参与临床试验的患者。虚拟临床试验的便利性有助于提高患者保留率,加快临床开发。图4为临床试验成本分布,其中患者招募占总成本的32%。AI/ML 应用可使临床试验更快、更安全、显著降低成本。AI 在改善患者体验方面的潜力,有助于在整个研发过程中更全面地融入以患者为中心的理念。
监管科学的加持,有助于增加AI/ML 应用在临床试验和药品研发全生命周期的权重。药品开发必须符合相关法律、法规的规定,同时以监管机构制定的相关指南、用例,作为行业在药品开发中运用AI/ML 的指针。药品监管机构认识到,利用数字信息整合研究与临床医疗照护,为研究开发和健康产出带来巨大的潜力,但应相应改革研究参与和数据共享的社会规范。以美国食品药品监督管理局(FDA)为例,该机构通过数字基础设施现代化促进、补充生物医药行业的工作,通过发展国家数字基础设施,以期实现迅速、系统地采集患者数据,与行业一起,共同了解医药产品在真实世界环境下的获益、风险。FDA 认识到,让消费者和患者直接参与FDA 的技术开发过程,成为利用社会科学和人类行为研究以及更有意义地与公众接触的一个重要机会[4]。
2023 年5 月,FDA 多个中心联合发布《人工智能和机器学习用于药品与生物制品开发的讨论文件》(Using Artificial Intelligence & Machine Learning in the Development of Drug & Biological Products :Discussion Paper and Request for Feedback), 就在药品、医疗器械(包括药械组合产品)开发中运用AI/ML 向利益攸关方征询建议、意见[5]。
2023 年8 月, 欧洲药品管理局(EMA) 发布《人工智能用于药品生命周期的观点文件》(Reflection Paper on the Use of Artificial Intelligence (AI) in the Medicinal Product Lifecycle)[6],阐述了EMA 对AI 支持安全有效地开发、监管、使用人用药品和动物用药品的看法。通过观点文件与开发机构、学术机构和其他监管机构展开对话,讨论相关领域未来发展方向,发展欧盟药品监管网络在数据驱动监管方面的能力,为这一快速发展的生态系统所带来的监管挑战做好准备,确保相关创新的全部潜力得以实现,造福患者和动物健康[7]。
本文基于FDA 讨论文件、EMA 观点文件,从药品监管的视角,浅析AI/ML 在药品临床试验中的运用,同时反映利益攸关方对相关主题的看法,以及可能面对的监管挑战。
1、 FDA 讨论文件中关于AI/ML 运用于临床试验的内容
FDA 讨论文件中关于AI/ML运用于临床试验的内容包括患者招募、试验参与者选择和分层、剂量/ 给药方案优化、参与临床试验患者的依从性、患者保持、临床试验场所选择、临床试验数据采集、管理和分析等方面,笔者已在《中国食品药品监管》杂志2023 年第7 期另文详细介绍[8],此处不再赘述。
2、 EMA 观点文件中关于AI/ML 运用于临床试验的内容
EMA 观点文件只关注AI 在药品生命周期中的使用。在临床试验中,会使用涉及AI/ML 技术的医疗设备,生成支持上市批准申请的证据,或能够与药品结合使用。在这种情况下,EMA 将参与评估相关设备的特性是否足以产生证据,以支持欧盟的上市批准。如果设备在产品特征总结中提供输入信息,例如在剂量学或监测方面,EMA 将评价拟组合使用的所有相关方面。
2.1 总体考量
EMA 认为,如果使用得当,AI/ML 工具可以有效支持药品生命周期内数据的获取、转换、分析和解释。模型引导的药品开发和生物统计领域的建议、最佳实践和既往经验也适用于AI/ML 领域。
2.2 药品临床试验标准规范
预期ICH E6《药物临床试验质量管理规范》中的所有要求将适用于临床试验中AI/ML 的使用。值得注意的是,如果AI/ML 模型是针对临床试验申请生成的,则完整模型架构、建模、验证和测试日志,训练数据和数据处理流水线的描述会被视为临床试验数据或试验方案案卷的一部分,因此应在上市批准或临床试验申请时,予以全面评价。在临床试验环境中应用AI/ML 时,需要考虑其他信息,其中对特定方面的影响,例如试验的复杂性水平、去中心化要素的使用以及作为决策支持软件的预期用途,应在具体方案的获益- 风险评价中予以反映。
2.3 临床试验中医疗设备和体外诊断试剂的使用
依据《欧盟医疗器械条例》(MDR)和《欧盟体外诊断医疗器械条例》(IVDR)监管医疗设备和体外诊断试剂[9-10]。AI/ML在药品开发和使用领域的应用,可能包括与此类设备的相互作用。当AI/ML 系统用于个体患者的临床管理时,根据MDR 或IVDR,可被视为医疗设备。这样的做法是出于完整性考虑,并不影响与医疗设备结合使用的药品的相关现行指南。关于MDR 和IVDR框架内软件的鉴定和分类的具体指南依据MDCG 2019-13[11]。根据上述规定对软件进行鉴定或分类,不在EMA 的职权范围内。使用CE 标志的设备,可能需要满足额外的要求,以获得在临床试验中使用的资质,以确保受试者的权利、安全、健康、数据的完整性和临床试验结果,包括相关设备的通用性。
2.4 数据分析与推断
AI/ML 模型用于药品临床试验中数据的转换或分析时,被视为统计分析的一部分,应遵循临床试验统计原则的适用指南,包括对下游统计推断影响的分析。在后期临床开发中,需要在统计分析计划中详细阐述预先指定的数据管理流水线和用于推断的完全冻结模型集。
2.5 早期临床试验
与药物发现类似,在临床开发早期阶段使用AI/ML 模型进行数据分析的风险通常很低,但可能包含影响患者安全的高风险应用,例如治疗入组或给药。在任何情况下,均应采取措施确保用于后续临床试验规划的所有估计在统计学方面是可靠的,同时应注意探索性分析与多重检验/ 多重性有关。对于早期临床试验的数据可能具有重大监管影响的情况,例如在有限的临床开发项目中,要求可能更高,应尽早与监管机构讨论交流。
2.6 关键性临床试验
在药品开发后期的关键性临床试验中,必须尽量降低与过拟合和数据泄露相关的所有风险。在部署相关模型之前,应使用前瞻性生成的数据测试性能,这些数据代表在预期使用场景的环境或人群中获取的数据。不接受增量学习方法,试验期间对模型的任何修改,均需要与监管机构交流互动,修改统计分析计划。
在打开用于假设检验的所有数据集之前,应以可追溯的方式锁定数据预处理流水线和所有模型,并在统计分析计划中记录。一旦打开数据集,对数据处理或模型的任何未预先指定的修改,都意味着分析结果被认为是事后的,因此不适用于生成验证性证据。如果可能,鼓励在关键性临床试验中部署模型之前,将其发布在开放存储库中。
2.7 精准医学
AI/ML 可用于根据疾病特征、患者基因型、宽带生物标志物组合和临床参数等开展的个体化治疗[4]。这可能包括患者选择、剂量、产品变体的全新设计以及从预先制备的变体库中选择。
有可能在产品特征小结中提及AI/ML 应用,以帮助作出适应症和剂量学方面的决定。在不影响其他监管机构开展合规性评价的情况下,药品的安全性、有效性以及AI 驱动的应用属于药品监管的范畴。
从药品监管的角度来看,在精准医学领域运用AI/ML 将被视为高风险应用,与患者风险和监管影响程度有关。除了针对高风险用例所阐述的原则外,还应特别注意界定剂量学变更,在实施前需要监管评价,制定处方者在实际运用中辩证领会的指南,包括发生技术失效情况下的后备治疗策略。
2.8 产品信息
用于起草、汇编、翻译或审查药品信息文件的AI 应用程序将在密切监控的环境下使用。鉴于生成式语言模型容易包含看似合理、实则错误的输出,在提交监管审查之前,需要建立质量审查机制,确保所有模型生成的文本所述事实、语法正确。
3、 利益攸关方对FDA 讨论文件的反馈
FDA 就讨论文件公开征询利益攸关方的建议、意见。截至2023 年11 月20 日,来自行业协会、生物制药行业、数字医疗、学者等利益攸关方的回复共计71份。囿于篇幅,本文选取有代表性的回复,简要介绍来自利益攸关方的相关看法、用例与具体实践。
3.1 国际制药工程协会的反馈
国际制药工程协会( International Society for Pharmaceutical Engineering,ISPE)希望强调模型选择、数据充分性(数量、质量、可见性、灵敏度等模型的重要性),重视数据管理活动(包括相关的验证和记录),定期审查(涵盖数据采集和数据管理),了解模型不确定性和预期的不确定性,以及不确定性的来源(例如模型参数的结构不确定性、残余误差的不确定性等)[12]。
ISPE 强调FDA 与其他监管机构保持一致的重要性。保持一致性将降低在药品生产中推进AI/ML 模型的障碍,有助于确保为患者供应优质药品。例如,对模型培训、持续验证、变更以及人为监督/ 受控程度的预期。推进AI/ML 在先进制药中的应用,需要国际协调。制药行业在很大程度上属于国际化行业,如果缺乏共同的监管方法,新技术实施会受限。
ISPE 认为,透明度包括明确说明AI/ML 系统在整个开发周期中如何学习、验证和更新,以及如何保持在受控状态,包括:性能和已知的故障模式;以可向客户解释的方式传达结果;应用特定AI/ML 技术的理由,与非AI/ML 方法相比的预期效益和风险。此外,透明度还意味着AI/ML 解决方案在多大程度上利用可解释的AI 技术,将所提供的结果或其他信息与专业领域专家对工艺过程的理解联系起来。
ISPE 认为,在药品开发过程中使用AI/ML 的主要障碍在于如何向受众解释AI/ML 系统在整个开发生命周期中如何学习、验证和更新,以及如何在整个开发生命周期中保持受控状态。这些模型一般无法分解成组成方程,因此,用户有赖于理解输入和输出之间的关系。此外,还必须对模型进行充分的多样性训练,以减少偏差。应制定适用的验证和报告结果的体系和规程。
ISPE 认为,基于风险、有意义的人为参与规范做法,人为参与程度应与AI/ML 模型的使用和风险成正比。人为参与程度也应与模型的不确定性相关。
ISPE 认为,可加强和实现可追溯性和可审计性的规程包括:模型版本化;模型与训练数据之间的联系;开发过程和见解的结构化说明,并与采用AI/ML 之前的原始认识进行比较;根据药品质量体系对模型进行定期审查;AI/ML 模型的开发可遵循ICHQ8/Q9/Q10 中所阐述的需要加以考量的要点,包括: 确定模型的目的;决定建模方法和实验/取样方法;选择变量/ 输入;了解模型的局限性和假设;支持模型开发的数据采集;开发和验证模型(可能有所重叠);评价模型的不确定性,并在存在需要的情况下,减轻模型不确定性的影响;用文件记录模型开发、验证以及生命周期验证计划;在药品质量体系中管理模型的维护和变更。
在药品开发过程中,AI/ML需要考量的数据质量、可靠性和代表性因素包括:数据生成时间,用于确定由于时间推移、对工艺过程理解的进步或数据生成过程变化而可能造成的偏移;将数据来源(内部、外部)作为代表性的相关驱动因素;源数据多样性和可变性,用于降低偏差风险。
ISPE 认为,在药品开发中使用AI/ML,有助于确保AI/ML 的完整性或解决偏差、数据缺失和其他数据质量的做法包括:数据去标识(视需要而定);数据使用的法律考量(例如检查与利益攸关方的合同);根据需要,在存储和传输过程中对数据加密;坚持适当访问数据的原则;采用联合学习方法,在多个分散的边缘设备或持有本地数据样本的服务器上训练算法,而不交换这些数据。联合学习使多个行为者能够在不共享数据的情况下建立一个共同的、强大的机器学习模型,从而能够解决关键问题,例如数据隐私权、数据安全、数据访问权和异质数据的访问[13]。
ISPE 认为,利益攸关方采用的有助于解决可重现性和可复制性问题的方法包括:带有时间戳的数据版本化;模型版本化;将模型版本与输入训练的确切数据点联系起来,包括训练期间的相关顺序;随机因素控制(例如,随机森林设置中的随机种子,包括如何选择适用种子或变更这类种子的政策);制药质量体系中强有力的变更控制。
开发者采用的识别确认和管理偏差的方法包括:从专业领域专家的角度评价数据集分层的性能;对各分层标准是否符合质量目标开展统计检验;在可行情况下,评价孤立因子和特征对预测可信度的影响。
对于模型的可解释性特征,例如采用图像识别技术的热图,ISPE 建议采用SHAP(SHapley Additive ex Planations)分解单个特征的预测效果;在分类设置中使用F1 分数以及与混淆矩阵相关的其他指标等关键绩效指标,或在点估计案例中使用平均绝对误差等指标;采用统计检验评价不同维度数据(例如时间或其他输入数据组) 的偏差;选择模型——在系统规范制定过程中确定最佳模型架构/ 算法的过程应记录在案并加以介绍;将可解释性扩展到围绕模型执行的推理代码,而不仅仅是模型本身。除其他软件外,开放源代码也可适用于模型(例如在部署迁移学习时)。所有系统都应完整记录所使用的代码/ 模型版本,并验证整个系统。
在数据源选择和纳入或排除标准方面,采用元数据捕获(例如数据捕获的时间点、源系统);从专业领域角度对用例进行业务分析,并将分析结果反映在专门用例的纳入或排除标准中,即说明理由和可理解的解释;专业领域专家、开发者和审核人员选择的多样性。
利益攸关方处理可解释性问题时, 采用与ICH Q8-Q11 一致的基于风险的方法,应为相关适用模型和系统设计决策提供信息;还应考虑到复杂性和可解释性。在决定是否采用可能在可解释性方面存在挑战的更复杂模型之前,应对用户群体进行全面了解。了解用户对可接受度的期望,以帮助从各种替代方案(从较简单到较复杂的模型)中选择,并确定人工-AI 团队的最佳解决方案。如果仅以统计学关键绩效指标来衡量,可能与最佳技术模型不同。
用于记录模型预测中的不确定性评价,以及如何传达不确定性的方法和标准,帮助支持不确定性评价。具体方法包括:根据对产品的理解和工艺过程知识,围绕点估计值或概率使用置信区间;设计专门的受控措施,包括估计值或概率及相关技术的不确定性,以及设计适用操作模式,例如,人在环中、人在环上或仅在例外情况下采取行动。定期监测模型性能统计关键绩效指标,并辅以更多定性手段,例如用户接受/ 拒绝和反馈。不确定性评价方法很多,ISPE 希望FDA 制定指南,阐述药品开发和生产过程中的不确定性近似方法。
3.2 美国普享药品协会的反馈
代表美国仿制药与生物类似药行业的美国普享药品协会(Association for Accessible Medicines,AAM) 指出,AI/ML 可用于仿制药和生物类似药开发。例如,AI 可用于开发体外-体内关系模型和预测,预测生物相关溶出方法和溶出曲线、预测生物等效性风险、选择规格以及受试药品和参比药品特性的相当性(例如粒度分布、溶出曲线)[14]。
AAM 表示,FDA 面临如何调整传统监管框架的关键问题,使得行业在将AI/ML 纳入药品开发计划时能够继续遵守监管要求。因此,必须从界定AI 规范工作入手,以便确定需要调整的监管框架活动范围。在讨论文件中,FDA 将AI 定义为“ 计算机科学、统计学和工程学的一个分支,使用算法或模型来执行任务并表现出学习、决策和预测等行为。” AAM 建议对此定义作出一些修改,以确保工作定义更准确地反映现有监管框架需要调整以适应AI 的新活动。AAM 建议对AI 的定义作如下修改:“计算机科学、统计学和工程学的一个分支,使用算法或模型来完成通常需要人类智能才能完成的任务和表现出通常需要人类智能才能表现出的行为, 例如学习、决策和预测。”AAM 建议加入“通常需要人类智能”这一表述,原因在于这是AI 的一个关键特征,便于将AI 与以前由人类完成的任务的简单自动化区分开。AAM 建议删除“学习”的提法,原因在于ML 只是AI 的一个子集,在其他地方也有定义,涉及不同的功能,在定义中加以区别会有帮助。采用这一替代定义可能有助于FDA 将监管框架的调整重点放在AI 所涵盖的活动上,这些活动需要不同的监管处理,并消除因使用应区别对待的更广泛活动而可能造成的混淆。按照同样的思路,AAM 还指出,讨论文件中的一些相关例子不符合AI/ML 的条件,而是属于使用技术和自动化的例子。例如,讨论文件中提到的“用于确保临床试验研究中坚持用药的工具”中,所列出的用于远程监控坚持用药情况的智能药盒和电子用药跟踪。AAM 认为,这些并不属于真正的AI/ML,只是对过去手工操作的活动进行技术/ 自动化,即使是用于触发不合规警报。
AAM 认为,讨论文件的重点是新药开发,但AI/ML 在仿制药和生物类似药的开发中也可发挥作用。FDA 应考虑制定一份单独的讨论文件,然后再制定一份指导文件,重点关注AI/ML 在上述领域的应用。例如,讨论文件可以涵盖仿制药和生物类似药开发中模型及其参数的选择,包括测试和训练数据集的选择、数据大小和质量,以及模型验证的要求。
AAM 表示, 用于AI/ML 的大数据集中的一些数据已经过时,而且患者很可能没有签署知情同意书,同意在AI/ML 应用中使用其数据。从监管机构的角度,这些数据是否可以使用;对如何在临床试验中使用这些数据有何预期;当AI/ML 预测患者有可能罹患某种疾病/ 病症时,在通知患者方面有哪些监管和伦理要求;AI/ML 使研究者和药品开发者能够识别可能的非结构化大数据集中的模式,在使用结构化和非结构化数据集时,有哪些监管要求;如何用通俗易懂的语言向试验参与者解释如何在临床试验中使用AI/ML ;当使用AI/ML 估算缺失数据值时,FDA 的预期是什么;AI/ML 填补空白与数据造假之间的界限在哪里;FDA 将如何对待那些被AI/ML 识别确认为有可能退出研究或带来其他挑战的潜在试验参与者(例如,有不依从史的参与者)被排除在外的研究计划,特别是考虑到增加少数群体代表性的规定;FDA 将如何对待那些被AI/ML 识别为有可能在研究期间发生不良事件的潜在试验参与者被排除在外的研究计划;FDA 将如何建议申办方平衡在反映一般人群的人群中研究药品的必要性,以及对已被确定为更有可能发生不良事件的患者的潜在伤害(AI/ML 预测的伤害比以前的方法更可靠)。
AAM 认为,对于在药品开发中采用AI/ML 的透明度,应被视为有关AI/ML 模型的适用信息,包括其使用、开发、性能和逻辑(如果存在)得到清晰传达的程度。
AAM 表示, 在仿制药开发过程中,AI/ML 还需要考虑其他数据。在仿制药开发过程中,生物等效性试验的数据集规模通常较小,模型可以不那么复杂,重点关注既往经验表明对确保仿制药与参比药品具有生物等效性非常重要的具体特征。再者,由于数据库规模较小,数据更有可能具有较高质量,模型也更透明。FDA 在完善仿制药开发中使用AI/ML 的思路时,应加以考虑。
3.3 英矽智能公司的反馈
对于以人为本的治理、可追责和透明度在AI/ML 药品开发中的具体用例或应用最需要哪些额外的监管清晰度,英矽智能公司认为AI/ML 的具体使用案例应涵盖分散式临床试验。分散式临床试验在为临床试验的设计和效率带来独特机遇的同时,也带来独特的挑战。例如,由于分散性,受试者依从性等问题可能会被放大。对于如何在非传统的研究配置或环境中使用AI/ML 采集不同地区数据,申办方可能需要更多的监管清晰度,以支持上市申请[15]。
英矽智能公司认为,在药品开发中使用AI/ML 时,为确保药品开发过程的安全性、准确性和符合伦理,人为参与不可或缺。相关的基于风险的做法应包括:①专业领域专家参与。AI/ML 专业领域专家参与整个AI/ML 开发过程,提供有价值的见解和领域知识。这些专家可以指导AI 模型的开发、验证和解释,确保其相关性和准确性。②利益攸关方应确保保存详细的AI/ML 开发过程文件记录,包括数据来源、预处理步骤、模型架构、超参数和决策依据等文档。此类文档可帮助监管机构和其他利益攸关方评价特定模型的可靠性以及所有潜在风险。③实施严格的验证和核查程序。实施以人为主导的严格确认和验证程序,以确保AI/ML 模型按预期运行并生成可靠结果。④使用可解释的AI/ML 模型。利益攸关方应优先使用可解释的AI/ML 模型。可解释的模型使专家更容易理解模型是如何生成具体决定或预测的,评价是否有必要进行调整,包括可能有助于保护临床试验参与者安全的调整。⑤利益攸关方应确保在药品开发过程中采用AI/ML 符合相关的伦理准则和法规,包括寻求伦理委员会的批准,尤其是在处理人体数据时。⑥利益攸关方应积极主动地发现和解决模型输出中可能存在的偏差和不公平性,避免出现意想不到的不良后果。根据金标准进行验证,将AI/ML 预测与既定的金标准或传统方法比较,为衡量准确性和安全性提供依据。要确定什么是金标准,就需要人的参与,特别是专家参与。例如,需要人工和专家的参与来选择最合适的指标,然后利用这些指标来评价AI/ML 预测结果。专家对数据质量评价的额外监督,可进一步确保准确性和安全性。为专家操作员实施一个强大的监控系统至关重要,以跟踪AI/ML 模型在真实世界场景中的表现。制定聚焦数据质量的内部规程。确保数据质量是AI/ML 应用成功的基础。利益攸关方应实施相关流程,以识别并解决可能影响AI/ML 模型性能的偏差、噪声数据和异常值。应考虑人因工程,以确保AI/ML 系统与药品开发工作流程完美契合。考虑可用性、用户培训和潜在的人为差错源。
对于数据质量、可靠性和代表性, 英矽智能公司表示, 数据、算力、新型 AI/ML 技术和高级分析技术的结合,使数据科学与生物学、化学和医学等更传统的学科成为药物研究关键核心学科。然而,迄今为止,在以连贯、稳健和系统的方式采集和协调临床试验终点和临床前数据方面进展有限。要充分发挥数据科学在药品研发中的潜力,就必须调整制药行业的结构和文化,使数据科学成为药品研发的核心学科。例如,建立数据科学团队并设立负责实施数据科学战略的高管职位。需要调动适当的财务资源,建立所需的底层架构,吸引并留住所需的技术熟练的数据科学家( 应在数据生成之前就参与进来)。这些步骤将有助于加快在行业内合理部署可靠的AI/ML 解决方案,也有助于推广执行FAIR [ 可查找(findable)、可访问(accessible)、可互操作(interoperable)、可重用(reusable)] 数据政策[16]。
开发者、制药商和其他利益攸关方一直在采用各种方法来确保AI/ML 的完整性,并解决药品开发中使用AI/ML 时出现的偏差、数据缺失和其他数据质量问题。例如,通过数据预处理和质量控制,可去除噪音、异常值和无关信息。此外,某些技术还能让利益攸关方减少用于训练AI/ML 模型的数据中的偏差。例如,重新取样(即AI/ML 和数据科学中使用的一系列技术,用于采集有关样本的更多信息,以提高总体准确性并更好地估计任何不确定性);数据扩增(即利用现有数据创建数据集的修改副本来增加训练集的规模,包括对数据集进行细微修改或使用深度学习技术生成新数据点);公平感知算法,这种算法在分析数据时会考虑潜在的公平性、歧视性、中立性和独立性问题。利益攸关方还可以利用LIME(本地可解释模型-不可知解释)或SHAP 等方法以及提供可解释性并为其预测提供解释的模型,深入了解模型决策。利益攸关方还应考虑验证模型的方法。例如,可以通过对各种数据集进行严格测试和评价来验证AI/ML 模型;可采用交叉验证技术来评价模型的可推广性和稳健性;还应将AI/ML 模型的预测结果与既定标准或专家进行比较,从而对AI/ML 模型进行外部验证。
3.4 美国注射剂协会的反馈
美国注射剂协会(Parenteral Drug Association,PDA)表示,在自动视觉检测和使用深度神经网络方面存在很大需求,与连续制造中使用的其他AI/ML 模型存在很大差异,难以解释。可能需要根据所使用的特定算法提供具体指导,例如模型的基本原理、使用该模型的预期结果。PDA 认为有必要就制定AI/ML 语言的整体开发、管理和受控计划提供指导,涉及临床试验的具体例子包括使用AI/ML 验证患者体验,确保临床试验期间所有受试者的平等代表性,从而利用临床研究管理规范确保药品按预期发挥作用。当在人体层面出现意外药物反应时,对于确证选择标准和受试人群分布,这一点尤为重要。
还需要考虑试验的多样性,例如从在一个国家开展试验转往与另一个国家一起开展试验。数据管理(例如数据清理、转换、数据准备)并不总是透明或可追溯的。算法选择、微调和模型创建是易于复制和评价的任务。以标准格式(例如开放神经网络交换)导出和封装AI 模型的能力是确保AI 模型避免像黑箱一样工作的推动力和促进因素。
PDA 提出的有助于确保AI/ML 的完整性或解决偏差、数据缺失和其他数据质量考量因素包括:在细胞基因疗法领域的临床研究中使用具备资质的患者材料/ 受试对象。AI/ML 有助于选择合格的患者,筛选出研究需要的特定细胞类型的患者,还可以根据需要的人群统计学随机选择合格的患者,确保所有性别、年龄、体重、身高、有其他易感因素者在研究中都有同等的代表性,并确保临床试验结果和安全性解释的顺利进行。AI/ML 还可用于根据研究标准确定受试者(符合资质的患者)在数据图中是否为离群值,并作为不将相关受试者数据纳入研究的正当理由。AI 开发者目前实施的主要做法完全符合现有的软件质量管理规范(AI= 软件+ 数据+ 计算)。此外,基于风险评价的GAMP5 v.2 是将其纳入AI 生命周期的一个非常好的方法,是将AI 风险评价客观化的一个很好的工具。在自动视觉检测环境中,数据质量是由技术专家的经验来保证的,他们了解变异的来源、缺陷的性质,以及既往关于偏差统计相关性的质量控制知识。迁移学习被认为是减少对稀缺产品大型数据集需求的一种有前途的方法,方法是迁移从类似产品(冻结深度神经网络模型的输入层)中获得的知识,并仅使用真实缺陷对模型的最后几层进行微调,以对分类进行微调。
PDA 认为,AI/ML 可帮助随机选择受试对象(符合资质的患者)的签号,以消除细胞基因疗法公司目前遇到的患者隐私权问题。对于自动视觉检测,大多数利益攸关方已决定在内部使用AI,而不将数据迁移出其设施。这对大型组织来说是可行的,但对小型企业来说,云端底层架构将是更好的选择,但需要考虑数据安全和隐私权。为了解决这个问题,通过将标注元数据(匿名化)排除在训练/ 验证/ 测试数据集之外来确保数据隐私权和安全性,以避免模型中包含隐私权信息的风险和偏差来源。
对于识别确认与管理偏差的方法,PDA 认为,AI 算法中的偏差问题在于,如果一定数量的受试对象都符合案例研究/ 临床试验标准,并且属于相同的人群统计标准。同样,如果较小的人群统计标准对临床试验有已确定的影响,则应考虑可能包括更多的代表性。AI 的目标是确保所有人群统计的平等代表性,包括指定的训练数据集作为程序,或特别标注风险,如果不能符合某些人群统计标准,必须成立专门的委员会来监督数据管理,以确保数据不出现偏差。此外,还有一些统计技术可用于测量和计算数据或样本中的偏差。迄今为止,都是基于AI 系统验证和维护期间产生的热图的人工解读。数据和元数据的紧密结合对于有效的数据管理至关重要。ALCOA+ 要求采用JSON 或XML 格式,但AI/ML 可以解释非人工可读的格式,因此需要制定使用AI/ML 格式的理由和解释策略。
对于针对特定使用环境的模型类型和算法选择,PDA 认为,使用决策树来确定选择正确算法的条件。例如,选择适用的估计器(例如scikit-learn);在自动视觉检测方面,使用最多的AI 模型是卷积神经网络,从最紧凑的VGG 到ImageNet,取决于目标硬件、数据集维度和所需的执行周期。确定何时使用特定方法验证模型并衡量特定使用环境下的性能(例如选择相关的成功标准和性能衡量标准),与临床和研发应用相关的考量因素,以及随后向生产流程的转移。根据不同的阶段- 关口流程,验证可能需要与患者评价的临床决策保持一致,以及在影响到生产流程的情况下。在描述预期用途后,必须立即定义相关的成功标准。
3.5 IQVIA 公司的反馈
IQVIA 公司表示,基于公平、透明、尊重、可追责和可审计性的原则,该公司认定几个与行业最佳实践基本原则密切相关或一致的主题:①以改善患者预后为使命。在未来几年中,医疗行业和监管机构将开拓新的领域,平衡AI 应用和决策方面的不确定性和前景,应立足于为患者提供更好的健康结局,优先采取行动。在审查AI 应用和监管要求时,提高患者医疗照护和福祉应成为行动指针,例如,引入公平原则以减少偏倚[17]。②与行业领跑者合作。行业与FDA 之间的持续合作,对于推动共同了解AI 在医疗保健和生命科学领域的潜在益处和局限至关重要。像IQVIA 公司这样积极参与AI 实际应用、方法开发/ 测试以及新型工具(例如基于GPT 的大型语言模型)开发的公司,可以为这一快速发展的领域提供真知灼见, 并为保持AI潜力与FDA 核心任务之间的一致性建立反馈回路。行业与FDA的AI 合作范围,应集中于当前受监管的药品开发流程和直接影响患者的医疗互联网技术应用,而不是扩展至更广泛的药物发现和运营效率活动。③透明度和可审计性标准。行业必须建立透明度标准,使利益攸关方和监管机构能够评价模型的有效性、可靠性和潜在偏差。在受监管环境下创建具有透明度的AI,必须能够被那些能够获取材料的利益攸关方和监管机构复制。过程透明会带来人对系统的信任。④利用现有框架和标准AI 术语。采用一致的AI 术语分类法,对于医疗保健领域内的有效沟通和理解至关重要。ISO/IEC 23053 等工作可为围绕AI 概念和应用建立共同语言提供详细而具体的基础。此外,IQVIA 公司强烈建议采用NISTAI 风险评价框架。该框架提供了一种基于原则的方法,贯穿整个AI 生命周期, 并与ISO/IEC23894 ∶ 2023 AI 风险管理指南保持一致[18]。该框架将成为评价医疗保健和生命科学领域AI 风险和受控措施的坚实基础。
IQVIA 公司认为,在药品开发过程中使用AI/ML 的透明度,即AI/ML 模型的相关信息公开传达给监管机构和利益攸关方的程度,包括有关模型开发、训练数据、算法、性能指标和逻辑,其预期用途和逻辑的详细信息,以及其预期用途和局限性。透明的AI/ML 系统能让监管机构和利益攸关方了解决策过程,评价模型的有效性、可靠性和潜在偏差。
为建立透明度,重要的是需要关注AI 系统的评价和验证过程,而非算法的“可解释性”。需要提供有关AI/ML 模型的使用、开发、性能以及根据当前实践对其进行验证和评价的方案的明确信息,包括定义成功的实证衡量标准,详细说明实验是如何建立的,并概述这些实验所使用的数据。建立用于衡量和验证AI 系统的方法的透明度,对于赢得信任至关重要。IQVIA 公司表示自身注重过程的透明度,使用户对AI系统产生信任。
IQVIA 公司认为,药品研发过程中实现应用AI/ML 透明度的主要障碍包括:知识产权问题、专属算法、商业竞争力、数据源的保密性以及AI/ML 模型的复杂性。使用AI/ML 会引发各种隐私权、监管和伦理问题,必须妥善处理。其他障碍包括:在验证方案和可接受性标准方面缺乏前期方案、数据质量问题以及缺乏接受AI/ML 模型的标准化指南。透明度的促进因素包括使用标准化的报告框架、利益攸关方之间的合作、采用开放科学原则以及明确的数据共享监管预期(在保护机密信息的范围内)。至关重要的是,要提前确定问题定义、实验设计和统计措施,让最终用户和专业领域专家参与到AI/ML 的开发过程中。开放数据平台、围绕基准和可重现性的分析、训练数据集的清晰度和说明以及共享代码也有助于提高透明度。数据共享可提高透明度,但在实践中也存在保密方面的挑战,例如与AI/ML 模型相关的知识产权问题和与患者数据相关的隐私权问题,这两方面都会阻碍透明度的提高。业界正在努力促进药品开发中更大程度的数据共享(例如通过制药行业软件用户交换组织和临床研究数据共享联盟),以及以标准化方式采用隐私权强化技术[ 例如《共享匿名和功能有效(SAFE)数据标准》(Sharing Anonymized and Functionally Effective (SAFE) Data Standard)][19]。随着保护机密和私人信息的行业标准不断发展,继续强调透明度仍然非常重要。
3.6 Verily 公司的反馈
Verily 是Alphabet 公司旗下的健康科技公司,专注于围绕精准医疗的研究、医疗照护和融资。Verily 公司表示,FDA 长期以来一直以透明的方式发挥着领导作用,确保上市销售的药品安全、有效。鉴于开发作为AI/ML 基础的工具和科学方法的多学科性质,特别是在药品开发方面,Verily公司对FDA 采取跨中心协作、协调FDA 机构内主要合作伙伴制定该框架表示赞赏[20]。
Verily 公司认为,精准医疗是利用来自各种临床和非临床来源的数据,为个人或群体提供最佳干预的健康方法。精准医疗代表着从“一刀切”向注重更全面的个人视角的医学范式的转化,这种转变带来了更加个体化的发展道路,包括使用AI/ML 选择和开发的新疗法。Verily 公司认为,AI/ML 有可能成为一种强有力的工具,在药物发现和开发活动整个过程中进一步释放精准医疗的前景。通过创建高质量、强有力和具有代表性的数据集,促进新疗法的开发,使干预措施更具针对性,更好地满足患者的个体化需求。此外,Verily 公司见证了利用AI/ML 改进临床研究(包括支持新型疗法注册审评的研究)的巨大潜力。利益攸关方将研究型企业整合到医疗体系中的势头日益高涨。这种整合可以加强研究者、患者和医疗服务提供方之间的合作。此外,目前正在开发新的分析方法、创新数据采集、整理和链接技术,借助这些技术,支持一系列研究设计,从而在非传统环境中开展研究。在更为宽松的环境中对患者开展研究,减少参与研究障碍。
在更宽松的环境中针对患者开展研究,可以减少参与障碍,确保更符合个人对医疗照护服务的偏好,并从整体上提供更相关、更具代表性、可操作性更强的证据。Verily 公司支持FDA 围绕旨在促进以患者为中心的更有力研究的指南所做的努力,包括使用创新数字医疗技术、应用真实世界数据,以及新颖研究设计,例如利用《药物临床试验质量管理规范》框架开展分散式临床试验。尽管取得这些进展,但在高效开展临床试验方面仍然存在障碍,尤其是在医疗照护点环境中实施时。医疗照护点试验提供了一种在大规模试验底层架构内采集临床数据的操作方法,可支持将临床研究与日常医疗照护服务相结合[21]。这样的方法需要多样化的数据采集技术,其中包含强大的方法论,以确保数据的质量、可靠性和代表性以及衍生证据的可信度,同时不会增加医疗服务的负担。Verily 公司认为,适当使用AI/ML 工具可以实现这些目标。
鉴于药品开发应用的风险程度不同, 应根据AI/ML 工具在支持药品开发和患者医疗照护方面的相关性对风险进行分类和具体说明。任何针对健康领域的AI 框架应在可行和相关的情况下, 与美国卫生及公众服务部(Department of Health & Human Services,HHS) 的优先事项保持一致,包括美国国立卫生研究院(National Institutesof Health,NIH)和美国国家卫生信息技术协调员办公室(Officeof the National Coordinator for Health Information Technology, ONC), 以及在HHS 以外可能在这些框架中发挥作用的机构, 包括美国国家标准与技术研究院(National Institute of Standards and Technology,NIST)等。
Verily 公司认为,AI/ML 在医疗保健领域,特别是药品开发领域的应用,可以在临床研究活动中为患者提供重要的保护,包括帮助使获得同意等流程更加稳健和以患者为中心。除了支持以患者为中心的同意程序外,Verily公司还认定AI/ML 应用于药品开发的优势,即为临床试验招募患者。在寻求招募多样化人群时,成本可能会更高,同时确保与研究临床相关的历史上代表性不足的人群充分参与。在招募和注册足够的相关研究参与者方面存在许多挑战。潜在参与者必须了解研究机会,有办法与研究进行沟通,表明参与的兴趣。试验招募推广工作通常依赖于传统渠道,例如与卫生系统和医疗服务提供机构的现有关系。AI/ML 可用于建立和分析各种数据集,帮助缩小研究与医疗之间的差距,如何在尊重患者隐私权的同时,更有效地识别潜在参与者和选择研究场所,公众对此非常关注。
Verily 公司还认为,AI/ML工具,特别是大型语言模型(large language model,LLM) 有机会更高效、更有效地提高参与者对研究的理解,这是招募和同意过程的一部分。例如,基于LLM的聊天机器人可以根据个人喜好,在不同的时间和环境下与参与者交流。作为精准健康试验方法的一部分,这些基于AI 的对话将来可用于支持试验人员在与参与者接触时提出更有针对性的问题。对于FDA 和更广泛的研究界来说,重要的是要考虑如何将LLM用作负责任的知情同意程序的一部分。
3.7 生物技术创新组织的反馈
代表生物制药行业的院外游说组织生物技术创新组织(Biotechnology Innovation Organization,BIO)支持FDA采取基于风险的审查、评价和监管方法,同时欢迎FDA 决定在现有指南和监管工具的基础上,在药品开发中使用AI,包括器械与放射卫生中心(Centerfor Devices and Radiological Health,CDRH) 指南和2021年发布的《用于医疗器械开发的机器学习质量规范指导原则》(Good Machine Learning Practice for Medical Device Development:Guiding Principles)[22]。BIO 认为,AI/ML 具有跨部门属性,因此,高层次的总体原则和标准应适用于所有部门,包括AI/ML 在药品开发中的应用。
BIO 认为,AI/ML 是一个充满活力的新兴领域,因此BIO 支持以渐进方式制定指南,让申办方和FDA 都有充分的学习机会。对于临床试验中的人体剂量,如果FDA 采用基于风险的监管框架,就必须制定明确的风险评价标准。还需要进一步明确在评价风险时应考虑的特征,例如发生概率、全身性和局部性风险的区别及时间框架。对于临床试验设计,监管部门最迫切需要明确规定临床开发AI/ML 用例, 这些用例可能对药品或生物制品的效益- 风险评价产生最大影响。需要进一步明确的具体用例包括用于患者识别、选择和分层的AI/ML 模型。需要明确风险评价在临床开发的不同阶段会有哪些不同,例如,Ⅰ期探索性研究与Ⅲ期注册研究的要求有何不同。具体到疗效评价,需要更多指南,说明FDA 需要哪些类型的证据来证明终点在未来的研究中是有效的。如果AI/ML 是在传统试验数据的基础上训练出来的,那么FDA 需要什么样的证据来证明终点在未来的研究中是有效的。如果申办方使用AI/ML 模型评价疗效,是否需要贴标? BIO 建议FDA 提供更多信息,说明使用依赖于AI/ML 组件的数字医疗技术来评价安全性和有效性的总体考量因素。
BIO 表示,需要明确医疗器械法规框架,特别是作为医疗器械的软件(SaMD)法规如何适用于AI/ML 应用。最好能举例说明临床研究中使用的不同类型的AI/ML,以及它们是否被归类为医疗器械。例如,如果AI/ML 应用于药品或生物制品开发的临床试验,但不打算商业化,也不影响患者的治疗或管理,那么应遵循哪些特定的器械法规,明确这些不同用例的验证和确认要求。
BIO 建议 FDA 继续向申办方和利益攸关方通报FDA 的现行AI/ML 相关项目和计划,建立透明、开放和协作的AI/ML 生态系统。另外,BIO 还建议FDA 开发一个决策树或快速入门指南,明确说明就哪些具体问题联系FDA的中心或团队(例如数字医疗卓越中心、器械与放射卫生中心等)。
对于数据可用性,BIO 认为,AI/ML 成功应用于药品开发依赖于大型数据库的可用性和可访问性,以训练系统。因此,访问政府发起的数据库将使从概念验证原型到真实世界技术的快速发展成为可能,从而加速生物医学研究。BIO 支持建立公立- 私营部门合作伙伴关系和其他合作关系,以推动用于药品开发的机器可读数据集的创建和共享。考虑到全球药品开发生态系统,BIO 建议FDA 考虑将美国以外的数据纳入现行或未来的合作中。
BIO 认为,数据质量属性应包括丰富的元数据或其他可能影响模型性能的技术。这些元数据将有助于提高模型的通用性,并避免因数据生成与训练集不同而产生问题。就数据集本身而言,需要足够大,以捕捉药品开发过程中的复杂性和可变性。此外,过小的数据集也有可能造成患者身份的重新识别。需要特别考虑使用小数据的特定案例,因为在这些案例中,预测或推论出现偏差的可能性要高得多。同样,作为特定人群样本采集的数据与总人群相比,也是一个重要的考量因素。
纵向或时间序列数据会给AI/ML 带来特殊的问题,目前的模型还不能自然适应这类数据。数据转换必须展示转换和传输数据的过程,以构成制药商用于运营的有意义的信息。由于通常很难理解使用不同数据源时的所有兼容性问题,因此在AI/ML 解决方案中使用异构数据源或组合不同数据源需要特别考虑。
对所有相关变量或特征的数据覆盖,偏差尤为重要,原因在于不包含所有相关变量或特征的数据可能会导致偏差或不准确的结果,这些结果可能无法完全解释或推广。数据集中的不平衡性,例如类别或罕见事件分布的变化,会影响AI/ML 模型的性能。要解决数据不平衡问题,需要采用超采样、欠采样或合成数据生成等技术,以确保公平地代表所有类别。
3.8 基因泰克公司的反馈
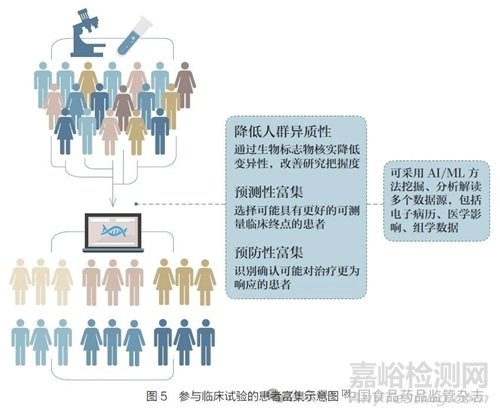
对于以人为本的治理、可追责和透明度,基因泰克公司认为,FDA 必须明确在哪些情况下,药品开发中使用的基于AI/ML 的数字工具会被视为医疗器械的软件。例如,在临床试验中,基于AI/ML 的工具可用于:根据指定的试验纳入或排除标准预筛或筛选患者;支持对不同研究臂的患者分层;富集试验中包括的特定生物标志物研究人群( 图5);选择纳入试验的患者。在这些情况下,进一步明确这些用例是否构成被视为医疗器械的算法,将使业界和临床试验机构明确适用的监管要求,以及在药品开发过程中使用这些算法所需的监管途径[23]。
基因泰克公司认为,在预测建模领域,必须就模型验证、可解释性和真实世界性能评价制定明确的指南。需要明确AI/ML 在患者选择、随机化和结果预测方面的使用规则。无论是在传统的临床试验过程中,还是在真实世界数据采集和证据生成的背景下,卫生部门对数据质量标准、算法透明度以及验证研究结果的方法和指标的要求仍然模糊不清。随着在自动化程度更高的环境中采用预测建模(即只需极少或无需人为参与),特别是在药品开发中,需要阐明与人工监督和透明度有关的标准、流程和程序。这种做法将与AI/ML 在其他环境中的适当和负责任的使用并行,以表明这种监督与该领域的大多数(如果不是全部)专家和组织保持一致。需要经过深思熟虑的指导,既包括总体监督,也包括对可能作为整体自动化设置的一部分而使用的特定模块的监督。
基因泰克公司认为,FDA 最好能明确定义透明度、可解释性。这两个术语经常被交替使用,尽管有所关联,但概念不同。这些模型的复杂性历来是一个障碍。ML 模型,尤其是深度学习网络,通常被视为黑箱,有时甚至被使用它们的科学家视为黑箱。ML 模型固有的复杂性会使其决策难以透明。
另一个障碍是非开源设置的模型和算法的专属性。出于对知识产权的考量,基因泰克公司可能不希望完全公开其模型和算法的细节,尤其是在这些模型和算法能推动重大开发的情况下。重要的是要区分AI/ML 在药品开发过程中不同环节的应用,例如临床试验、安全性评价、药动学/药效学。这些用途中,有很多可能被视为商业秘密,给申办方带来了巨大挑战,既要满足监管机构和社会对透明度的需求,又要保持适用数据保护。此外,在AI/ML 模型的报告/ 披露方面,缺乏广泛接受的标准或指南,这可能会导致申办方在哪些信息需要共享、向谁共享、以何种格式共享以及在何种情况下共享等方面的不一致和困惑。
最后,数据隐私权和数据传输也是潜在障碍。为适当训练AI/ML 模型所需的数据量,往往需要使用全球范围内的数据。这可能会受到特定司法管辖区数据传输要求的阻碍。透明度要求清晰沟通用于模型训练的数据,但有可能与隐私权法规和专有数据问题相冲突,特别是在生物制药的全球环境中,敏感数据成为常态。
尽管存在障碍,但在如何提高透明度方面正在取得进展,其中可能包括可解释的AI/ML 模型,开发更多可解释的模型或复杂模型的可解释性技术本身,能够促进透明度。开源和AI/ML 开发的各个方面也有可能促进透明度。学术界开放源代码和预印本的趋势使研究更加透明,促进了思想市场的竞争。产业界、学术界和监管机构之间的合作,可以通过就最佳实践达成共识来促进透明度。
罗氏公司与斯坦福大学合作开展了一项研究, 开发出TrialPathfinder 模型,用于系统评价包容性研究背景下的纳入或排除标准及其对试验入组的影响[24-25]。此外,罗氏公司还开发了一种新颖的指标R 指数,用于总结临床试验与基线人群相比的代表性,以便对数据代表性进行比较和设定基准。
3.9 美国药品研究与制造组织的反馈
代表制药行业的院外游说团体美国药品研究与制造组织(Pharmaceutical Research and Manufacturers of America,PhRMA) 表示,AI/ML 是临床试验的一个关键因素,而相关临床试验是药品获批的依据,所需的详细程度和文件要求将更加明确[26]。可能包括下述情况:AI/ML 用于初步分析(例如协变量调整)、初步分析结果评估(例如使用算法替代人工读数)、关键性试验关键数据集的清理、正在开展关键试验安全性数据的监测、基于AI 的终点的使用、人群药动学/ 药效学建模以及试验目标人群选择。
PhRMA 认为,在药品开发过程中使用AI/ML 系统的常见透明度障碍之一,包括用于训练、验证和解释复杂模型的大型、定义明确且具有代表性的临床试验数据集可用性方面存在局限。从监管角度看,必要的透明度水平应基于风险,并取决于特定的受众和使用环境。例如,对于在临床试验中使用某些AI 系统,或对于作为新药申请或生物制品许可申请的一部分提交的数据包中使用某些AI 系统,FDA 的透明度要求,可能不同于针对医疗照护专业人员的透明度要求。
4、 结语
在所有工业门类中,针对制药工业的监管强度,仅次于核工业。临床试验对确保药品的安全性、有效性至关重要。药品、生物制品能否获得监管机构批准,取决于临床试验(尤其是晚期临床试验)生成的证据。一方面,随着监管机构加速批准(附条件批准)的药品越来越多,上市后临床试验和纵向观察性研究越来越重要。另一方面,以肿瘤药为例,获批新药针对的适应症人群越来越小,所针对疾病的亚型越来越细。一些畅销药品,在最初作为罕见病用药获批后,采取扩展适应症或改变用途、重新定位策略,逐渐扩展适应症,增加销量。AI/ML 可以在所有这些过程中发挥重要作用,提高临床试验效率,降低成本。AI/ML 本身具有跨学科、跨平台运用的特点,在肯定AI/ML 运用于临床试验带来乐观预期的同时,也应看到所面临的监管挑战。例如,如何进一步明确AI/ML 在制药行业尤其是临床阶段中监管部门的责任和权限;制定严格的数据信息共享规则;确保患者的隐私权安全保障。同时考量由于AI/ML 版本模型快速迭代、数据增量以及其他技术进步所带来的监管不确定性。因此确保AI/ML 运用与包括临床试验在内的药品、生物制品全生命周期开发稳健可靠发展,需要药品监管机构与利益攸关方沟通、协作,制定、完善相关的行业指南、用例。