导语:临床评价可谓设计确认过程中的重要环节,也是贯穿医疗器械全生命周期的以确认医疗器械在其适用范围下的安全性、临床性能和/或有效性的持续进行的活动,同时也是影响医疗器械开发成本的重要一环,因此如何正确选择临床评价路径对于器械研发人至关重要。本文着重介绍临床评价与设计开发的关系及在各阶段的任务、临床评价流程及路径、医疗器械临床评价路径选择以及临床评价涉及到的法规,以为诸君在研发过程中提供点滴参考。
第一部分 临床评价与设计开发的关系
临床评价选择的重要性无容置疑,那么我们在设计开发的各个阶段应该做些什么呢?
立项和策划阶段:应明确产品的目标市场,对产品分类、注册单元、注册路径、上市前临床路径进行策划,输出“注册/临床路径”。
设计输入阶段:根据产品注册与临床策划时确定的产品目标市场和产品分类,识别该产品应遵循和部分遵循的规定产品技术要求和规定产品实现过程要求的法规和标准。比如,是否存在相应的临床评价注册指导原则或同品种临床评价注册审查指导等。若采用同品种对比,则应对选择的对比器械进行设计输入。
设计输出阶段:当产品为免临床目录内产品或通过同品种比对豁免临床试验时,应进行产品与对比产品性能对比评价。该评价应至少包括:目的、检验设备要求、样品信息、对比项目及检验方法、测试结果、结果分析等。该评价应在设计输出评审完成前完成。产品与对比产品性能对比评价结果是产品性能及指标制定依据的输入来源。
当进行创新产品时,在设计定型阶段必要时进行早期小样本临床。前期撰写的文章医疗器械研发能否开展小样本临床试验中FDA发布的《Investigational Device Exemptions (IDEs) for Early Feasibility Medical Device Clinical Studies,Including Certain First in Human (FIH) Studies》(简称EFS)介绍到:早期可行性研究允许对器械进行早期临床评估,以提供原理和初步证据临床安全数据。这些研究对于临床早期器械开发经验是必要的,因为非临床测试方法不可用或不足以提供推进开发过程所需的信息。
设计验证阶段:对于采用同品种等同性临床评价的,动物试验往往也是对于技术特征等同性的证据之一。同时若采用临床试验,则该阶段应进行相应的临床试验策划,如选定机构、临床试验方案等等。
设计确认阶段:这一部分在浅谈设计确认开展及输出物要求(无源医疗器械详细攻略)进行了详细的介绍,这里简单概括如下:
A 临床试验策划需要开展临床试验的产品,应输出临床试验决策。本阶段应进行临床试验策划,策划活动包括临床试验审批/备案资料准备、伦理资料清单及提交资料、临床试验机构的筛选等,伦理批件应在临床试验入组前获得。需要进行临床试验审批的器械详见《需进行临床试验审批的第三类医疗器械目录》(2020年修订版)(公众号内回复 临床审批可获得)。
对于需要临床试验的产品,应制定临床试验方案以满足设计确认的要求。适用时,本阶段应输出临床试验的设计依据。临床试验方案的设计应遵循食品药品监管总局2018年第6号通告《医疗器械临床试验设计指导原则》的要求。完成设计确认活动,更新设计确认追溯表,应包含设计确认的结果、结论,并记录任何必要的措施。
B 临床试验样品
适用时,应出具临床试验用样品生产方案,且应经评审,通过后方可进行(由于临床试验费用是比较高的,因此建议对于临床样品的生产方案进行评审)。需要开展临床试验的产品,本阶段依据设计定型评审后文件版本进行制造,保留完整的生产和检验可追溯性记录。临床试验样品应保留完整的出入库记录、去向表。
用于临床的应保留储运记录、回收记录[医疗器械注册质量管理体系核查指南(2022年第50号)规定申请人应保存临床试验产品的分发、储运、回收/退回等记录]。临床首批样品入库前完成临床首批样品入库前体系自查,输出核查报告,完成不符合项整改。由于临床时期存在一定的时间空档,此时可以利用临床试验的时间空档整改不符合项,便于后期一旦获得临床报告后即可提交注册。
C 临床评价
需要开展临床试验的产品:本阶段应根据临床试验方案实施临床试验以满足设计确认的要求,应输出临床试验报告和统计分析报告。在注册提交前应输出临床评价报告,具体应遵循《国家药监局发布的医疗器械临床评价技术指导原则(2021年第73号)》医疗器械注册申报临床评价报告技术指导原则的要求。
无需临床试验的产品:详见免于临床评价医疗器械目录的通告(2023年第33号)(回复 免临床 可获得),本阶段应输出产品对比说明。具体应遵循《国家药监局发布的医疗器械临床评价技术指导原则(2021年第73号)》列入免于临床评价医疗器械目录产品对比说明技术指导原则要求(公众号内 回复 免临床 可获得)进行。
同品种等同性论证的产品:对于需要进行临床评价的第二类、第三类医疗器械,若通过等同器械的临床数据进行临床评价,需要进行等同性论证并输出报告,具体可参照《医疗器械临床评价等同性论证技术指导原则》(公众号内 回复 等同性 可获得)进行。
第二部分 临床评价过程及路径
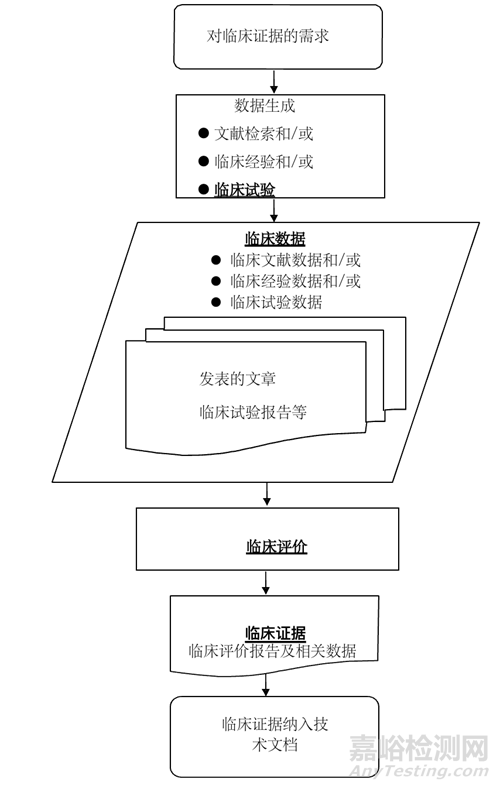
在《医疗器械临床评价技术指导原则》中介绍过临床评价的流程,如下图所示:
临床评价的结果为临床评价报告,是对临床数据及其质量进行详细阐述,论证临床数据如何证明产品对安全和性能基本原则的符合性。临床评价需持续开展,产品上市后注册申请人需对产品安全性、临床性能和/或有效性信息进行常规监测,并根据更新的信息,进行风险受益再评估。临床评价的输入主要是来源于临床试验报告、临床文献和临床经验的临床数据。根据产品特征、适用范围、注册申请人宣称、警示及注意事项的充分性、临床使用经验的不同,论证产品对安全和性能基本原则符合性需要的临床数据和证据亦不相同。临床评价旨在证明与患者受益相比,产品使用相关的风险可接受,且能较高程度地保护患者健康及安全。因此临床评价需与风险管理文件相互参照。
医疗器械临床评价技术指导原则
根据国家药监局关于发布医疗器械临床评价技术指导原则等5项技术指导原则的通告(2021年第73号)我们可以得知,临床评价的路径主要包括:免临床评价(简称免临床)、临床试验和同品种等同性论证(简称同品种)三种路径。
第三部分 临床评价路径选择
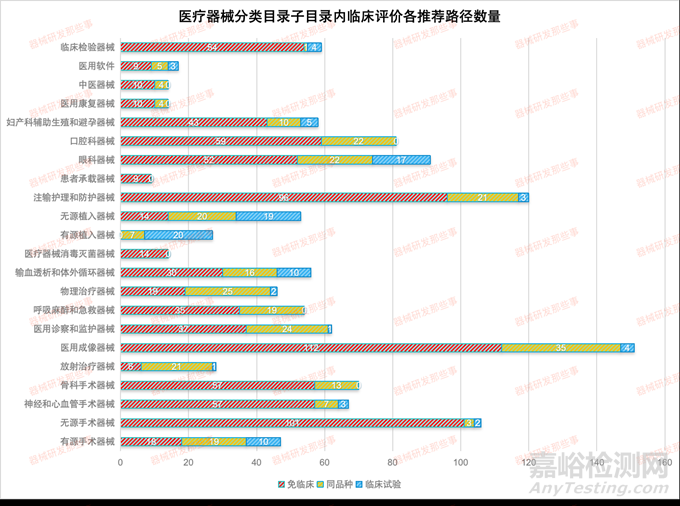
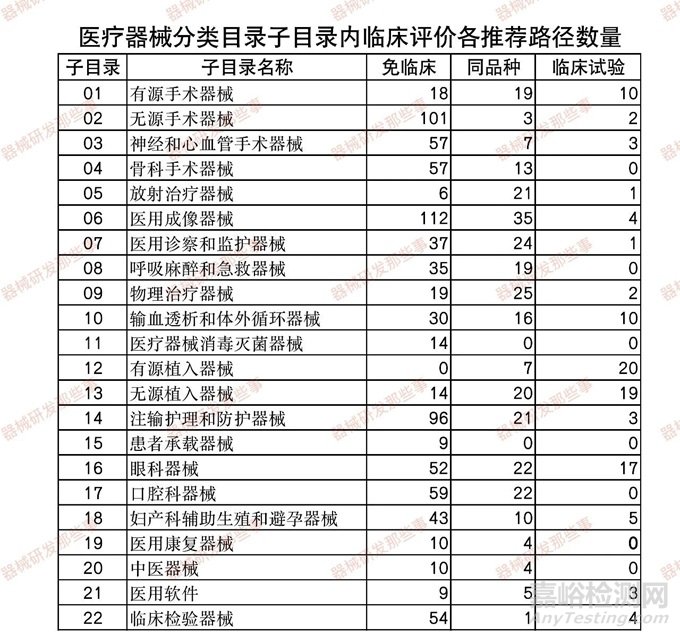
根据CMDE发布的《医疗器械分类目录》子目录01-22相关产品临床评价推荐路径的通告(2022年第20、24和30号) 相关内容,统计了在各个子目录下的产品对应推荐的临床评价路径的数量(免临床评价-简称免临床、临床试验和同品种),如下表和下图所示。
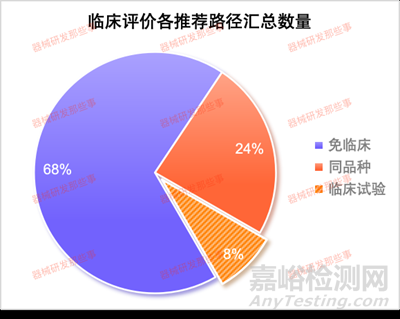
从图中可以看出,涉及到临床试验的子目录相关产品占比8%,免临床评价的占比68%,同品种对比的占比24%。其中涉及临床试验的主要集中在12有源植入器械、13无源植入器械和16眼科器械以及01有源手术器械。
第四部分 临床评价涉及的法规
《医疗器械监督管理条例》(2020年12月21日)
《医疗器械注册与备案管理办法》(国家市场监督管理总局令第47号)
《医疗器械临床试验质量管理规范》(国家药品监督管理局国家卫生健康委员会2022年28号公告)
国家药监局关于实施《医疗器械临床试验质量管理规范》有关事项的临床文献数据通告(国家药品监督管理局通告2022年第21号)
《医疗器械临床试验设计指导原则》(2018年第6号)
《接受医疗器械境外临床试验数据指导原则》(2018年第13号)
《真实世界数据用于医疗器械临床评价技术指导原则(试行)》
(2020年第77号)《医疗器械拓展性临床试验管理规定(试行)》(公告2020年第41号)
《需进行临床试验审批的第三类医疗器械目录》(2020年修订版,通告2020年第61号)
《免于临床评价医疗器械目录》的通告(2021年第71号)
《医疗器械临床评价技术指导原则》(2021年第73号通告)
《决策是否开展医疗器械临床试验技术指导原则》(2021年第73号通告)
《医疗器械临床评价等同性论证技术指导原则》(2021年第73号通告)
《医疗器械注册申报临床评价报告技术指导原则》(2021年第73号通告)
《列入免于临床评价医疗器械目录产品对比说明技术导原则》(2021年第73号通告)
《医疗器械临床试验数据递交要求注册审查指导原则》(2021年第91号)
《医疗器械真实世界研究设计和统计分析注册审查指导原则》(2024年第3号)
结束语:临床评价在设计开发过程中重要性无容置疑,本文详细介绍了临床评价与设计开发的关系以及各阶段应进行的与之相关的事项、临床评价路径及流程、临床评价推荐路径以及涉及到法规。