非临床动物研究中最常用的4种给药途径是静脉(Intravenous, IV)、皮下(subcutaneous, SC)、腹腔(intraperitoneal, IP)和口服(Oral)。尤其啮齿类动物试验中,IP是非常常见的。IP的优势包括给药方便、快速、动物应激反应轻、给药体积大(高达10mL/kg)等。当然,IP也有劣势,经常被质疑和挑战的点聚焦在与临床拟用给药途径不一致。因此,有必要对腹腔的解剖学和生理学特点、IP给药后的吸收机制等进行总结,厘清动物IP给药的一些误区。
腹膜腔的解剖学和生理学特点
腹膜腔来源于胚胎的体腔,是腹部位置的紧密空间,内含多个腹部器官。腹膜腔内分布有广泛的膜比如腹膜,其总面积与皮肤面积相当。腹膜腔内有一层由水、电解质、蛋白质、细胞和组织间质液组成的液体薄膜,即腹膜液。人体中,腹膜液体积约50-75mL,小鼠中约为0.02-0.1mL。此外,腹膜液中还含有白细胞、抗体和血浆蛋白。
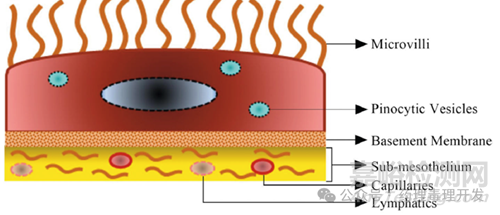
腹膜由单层鳞状间皮细胞组成。间皮细胞层位于一层薄的基底膜上,大多数间皮细胞呈扁平型,直径约为25μm。间皮细胞通过紧密连接、粘附连接、间隙连接或桥粒相互紧密连接。腹膜的间皮下层含有胶原蛋白、脂肪组织、淋巴细胞、血管和淋巴管。间皮细胞的顶端表面含有不同长度、形状和密度的微绒毛,这些微绒毛增加了腹膜的功能表面积。
腹膜间皮细胞在维持腹膜稳态以及液体和溶质跨膜运输中起着至关重要的作用。腹腔内器官和肠系膜均需要内脏腹膜的支持。腹膜可最大限度地减少摩擦,促进腹部内脏之间的自由运动,抵抗感染或定位感染部位,并储存脂肪,尤其是在大网膜中。腹膜间皮细胞具有很多上皮细胞的特征,包括具有表面微绒毛、极化成单层细胞并允许分子单层运输。腹膜液的pH值为7.5–8.0,具有缓冲能力,因此IP给药后腹膜腔内的物质很少电离。
腹腔血流
腹膜的间皮下层有一个复杂但有效的血液和淋巴管网络。其血管类型以细血管为主,也可见少量小动脉和小静脉。间皮下层的毛细管分布密度沿着腹腔不同位置而变化。比如,在兔中,腹膜腔血管最丰富的区域是肠系膜,包含了71%的腹腔毛细血管。在兔和大鼠中,通过腹膜腔中的毛细血管可以接收到4-7%的心脏血液输出量。大鼠中的平均腹腔血流速度是2.5-6.2mL/min/kg。另外,内脏腹膜和腹膜内器官的血液供应来源于腹腔、肠系膜上动脉和下动脉,而壁腹膜则由旋髂动脉、腰动脉、肋间动脉和上腹部动脉的分支灌注。同样的,从内脏腹膜排出的静脉血流入门静脉,而从壁腹膜排出的血管流入下腔静脉。总的来说,整个腹膜都充满了毛细血管,并为腹膜腔和血浆之间的药物交换提供了充分的接触面。
腹腔淋巴系统
与其他器官类似,腹膜内也含有淋巴网络,为腹膜组织的溶质和液体交换提供支持,防止水肿的形成。在腹膜的间皮下层,末端淋巴毛细管细分为胸骨旁、椎旁、纵隔、肋间和腹膜后淋巴管。胸骨旁、纵隔和腹膜后淋巴管分别位于横膈膜的前部、后部和中部。这些末端淋巴毛细管汇合在一起形成收集和结前淋巴管,然后与区域淋巴结连接。胸骨旁、椎旁和纵隔淋巴管与纵隔淋巴结相连,而腹膜后淋巴管与脑池和肠淋巴结相连。从这些淋巴结,淋巴系统通过胸管和右淋巴管与静脉循环系统相连。
溶质在腹腔的吸收
腹腔因其表面积大(如大鼠腹膜面积约125cm2)、间皮细胞存在微绒毛、血供大有利于快速吸收等特点,是物质经IP给药后进入系统循环的优良路径。此外,淋巴转运也是物质从腹膜进入系统循环的一大助力。
不过,IP给药后的物质在到达血管之前需要经过若干道屏障,包括腹膜液、间皮细胞、间皮下层及血管壁。内脏和壁间皮中的跨细胞和细胞间间隙是溶质和分子从腹膜腔进入周围组织的主要通道。由于不同结构特点,与壁间皮相比,内脏间皮对分子的渗透性更强。从解剖学上讲,内脏腹膜由扁平细胞组成,细胞内有大量的pinocytic vesicles,有助于分子的吸收。相反,壁腹膜含有较少的pinocytic vesicles,具有更发达的基底膜/结缔组织,这使其对分子的渗透性较低。如下图蓝色小泡所示。
实验研究表明,中、小分子量的物质(MW<5000)和液体主要通过脾脏、肠系膜下和肠系膜上毛细血管的扩散从内脏腹膜吸收,并进入门静脉。另一方面,大分子(MW>5000)物质、蛋白质、血液和免疫细胞被淋巴管吸收。如下图所示,线的粗细代表主要吸收路径。这点其实与皮下吸收路径有些类似,分子量越大,淋巴吸收占比越高。
流体或者其他物质从腹腔入血或者从血回到腹腔的机制是相似的,均是通过扩散或者对流。小分子从腹腔入血的吸收速度主要取决于3个因素:有效表面积(A)、溶质浓度(ΔC)和溶质的渗透性(Ps),公示如下:
简言之,有效表面积越大(主要指间皮细胞绒毛面积)、浓度越高、渗透性越强(如溶质的脂溶性高),吸收速度通常越快。
IP给药后的毛细血管吸收
腹膜毛细血管的毛细血管壁为“连续”型,内衬单层连续内皮细胞和基底层。这些内皮细胞非常薄(0.5μm),具有高度渗透性,此外还含有大量细胞质囊泡。位于相邻内皮细胞之间的6-7 nm狭窄细胞间隙也存在于这些毛细血管中,确保水溶性离子和小分子的快速通过。分子量<20000的物质可以通过扩散从腹腔吸收入血,扩散的速度和程度取决于分子的大小、电荷、构型和浓度梯度。此外,溶液形式的药物IP给药后增加了腹腔内静压,促使可溶性药物与液体一起通过腹膜对流进入毛细血管。从内脏腹膜、肠系膜和网膜吸收的分子流入门静脉,而从壁腹膜毛细血管和淋巴管吸收的分子则直接进入系统循环。通过门脉循环进入的药物在通过肝脏后与体循环合并,会引起药物的快速代谢。
在一项对大鼠中进行的研究中,Lukas及其同事表明,IP给药小分子(阿托品、咖啡因、葡萄糖、甘氨酸和黄体酮)后,主要的吸收途径是通过门静脉循环。此外,从吸收速度对比看,IP是比SC快的,IP给药后最快10s即可在系统循环中检测的到药物,SC则需要60s。但SC肝脏暴露程度较IP低,因此被肝脏转化、代谢的比例低。
影响化合物IP吸收的另外一个因素是脂溶性。通常,脂溶性增加腹腔吸收随之增加。比如,大鼠腹腔注射巴比妥后,仅有约57.4%的巴比妥被吸收(脂水分配系数为0.001),而硫喷妥钠的吸收(脂水分配系数为3.3)为96.1%。此外,解离状态和粘度也是影响IP吸收的重要因素。一般来讲,非解离化合物、制剂粘度低的化合物,更利于IP吸收。
IP给药后的淋巴吸收
分子量超过30000的大分子腹腔注射后,主要通过淋巴管吸收入血。除了蛋白外,小的颗粒和细胞也主要通过淋巴管吸收。值得一提的是,呼吸过程中横膈膜的松弛也影响大分子从腹腔的吸收。呼气时膈肌松弛,陷窝边缘相邻的间皮细胞相互分离,产生吸力,促进大分子的吸收。大约75%的被吸收的蛋白质流入右淋巴管,25%流入胸导管,随后与静脉循环汇合。值得注意的是,右淋巴管和胸导管的阻塞并不妨碍蛋白质从腹腔的系统吸收,因为微量蛋白质仍然可以通过其他小的淋巴-静脉互通和毛细血管壁吸收。大分子IP给药后淋巴吸收过程如下图所示。
小分子IP给药vs其它途径给药
Durk及其同事比较了卡马西平、西酞普兰、去甲基氯氮平、苯海拉明、加巴喷丁、美托洛胺、纳曲酮、奎尼丁和利培酮9个分子的IP和SC给药药代动力学参数。结果发现,所有分子IP给药后的达峰更快,峰浓度也更高。另外,大部分化合物IP和SC给药的脑部暴露量高于IV。IP与口服相比呢,以多西他赛(分子量807.89)为例,口服和IP的绝对生物利用度分别为2.8%、69%。以deramciclane(分子量301.466)为例,IP的绝对生物利用度是口服的约6倍(18.5% vs 3.4%),达峰时间比口服快4倍。
总之,与SC和口服给药相比,小分子IP给药后的吸收速度更快,程度也更高。
大分子IP给药vs其它途径给药
IP是生物药非临床研究中常用的给药途径,尤其是药效学试验。那么与其他给药途径相比,IP的优势是什么呢?
有团队研究了TNF受体-IgG融合蛋白(分子量约210KDa)IP、SC和IV给药后的外周暴露量情况,发现达峰浓度和AUC从高到低依次是IV、IP和SC。其中,IP的AUC是SC给药途经的5.5倍。
重组人红细胞生成素(分子量约37KDa)以5000U/kg,IP和SC给予大鼠后的AUC分别为140331、117677 U.h/L、Cmax分别为10015、6224U/L,Tmax分别为3、9h。
GLP-1受体激动剂艾塞那肽(分子量约4186.63)给予大鼠50nmol后,IV、IP、SC对应的AUC分别为172、128、112 nM.h/mL。IP和SC的Cmax分别为35.3和28nM。不过,IP消除较SC更快一些,t1/2分别为157、216分钟。
抗人CXCR4单域抗体(分子量约60.6KDa),以10mg/kg剂量给予小鼠,IV、IP和SC对应的AUClast分别为1871、1435、760μg.h/mL,Cmax分别为467、176、32.17μg/mL,Tmax分别为1.8min、2h、8h。IP较SC的暴露量更高,达峰更快。
CD20抗体veltuzumab(分子量约145KDa),以150μg剂量给予小鼠,IP、SC对应的AUC分别为106.639、51.67nmol.h/mL,Cmax分别为0.195、0.203nmol/mL,两种给药途径的Tmax均为24h左右。IP对应的AUC更高,Cmax和Tmax二者相当。
整体看,对于大分子生物药来讲,IP的吸收程度如AUC是优于SC的。
混悬液IP给药的生物利用度
混悬剂腹腔给药后主要通过淋巴系统吸收,吸收主要受药物的理化性质、溶出速率和粒径大小影响。比如6-甲基香豆素(不溶于水,分子量约160.17)以200mg/kg的混悬剂形式,经IP、PO给予大鼠后,对应的AUC分别为2177.0、977.2μg.min/ml,Tmax分别为6、30min,无论吸收速度还是程度,IP均表现更好。又如elacridar,当以羟丙基甲基纤维素和吐温-80制备成混悬液后,IP给药的AUC远低于口服(90.3 vs 1460),当以Cremophor EL, Carbiton和Captex 355制备成微乳给药后,IP给药的AUC反而明显高于口服(962 vs 270),原因与混悬剂的溶出速率明显慢于微乳有关。有研究显示,不同粒径的戊巴比妥(<44 μm和297–420 μm)经IP给予小鼠后,小粒径的药物毒性明显高于大粒径。
IP给药的局限性
首先,前文其实已有提及,IP给药后有些药物,尤其小分子,经腹膜吸收入门静脉,进入肝脏,是有首过效应的。生物药IP给药后经淋巴管吸收,反而受首过效应影响较小。其次,无菌和无刺激性对于IP注射给药的药物也很关键。有刺激性的化合物可能会引起肠梗阻或腹膜炎症,并有进一步发展成组织黏连的可能。另外,约20%的IP给药会出现给到皮下的情况,主要原因与给药角度过小有关。IP给药如果体积过大,比如大鼠给药体积>10mL/kg,会出现导致疼痛、化学性腹膜炎、组织纤维化、腹部器官穿孔、出血和呼吸窘迫等症状。最后,反复IP给药会导致刺激作用累积,且反复针刺也会引起腹膜损伤。当然,IP、SC、口服、IV不同给药途径各有其优缺点,具体如下表所示。
最后
IV是生物利用度最高的给药途径,但不是所有药物均可溶于水或形成溶液状态、小鼠血管小从而给药难度大、IV不适合长期/反复注射,口服、IP、SC等给药途径是不错的备选。相较于其它给药途经,IP具备给药方便,易于操作,大小分子均可实现不错的生物利用度,吸收速度和程度通常优于肌肉、皮下和口服给药途径,溶液和混悬液/乳剂均适用等特点,使IP成为非临床研究中的常用选择。IP给药后主要有两种吸收路径:1)通过内脏腹膜、肠系膜和网膜吸收,之后进入门静脉循环;2)通过壁腹膜和淋巴管吸收,直接绕过肝脏进入体循环。一般,小分子通过路径1)入血,大分子通过路径2)吸收。当然,对于支持注册申报的毒理学研究,还是建议采用临床拟用途径。对于啮齿类药效学研究、早期靶点验证研究等,IP给药其实是个不错的选择。