药品研发实验记录是指在药品研发过程中,应用实验、观察、调查或资料分析等方法,根据实验实际情况直接记录或统计形成的各种数据、文字、图表、照片等原始资料。应规范管理药品研发实验记录,保证研发实验记录、数据的真实、准确、完整、可追溯。
众所周知,只有通过官方的技术审查、研制和生产现场核查,产品才能获得注册批准,国家药品监督管理局食品药品审核查验中心在2021年12月20日发布了《药品注册核查要点与判定原则(药学研制和生产现场)(试行)》,自2022年1月1日起施行;其中药学研制现场核查是通过对研究过程中原始记录、数据及现场进行核实和/或实地确认,核实相关申报资料的真实性、一致性。
而药学研发实验记录是撰写药品申报资料的依据,申报资料中总结或提炼的实验所使用的物料、仪器设备,采用的实验条件、实验方法、操作步骤、实验过程,观察到的现象,测定的数据,得出的结果结论等均在药学研发实验记录中有记载和体现。只有规范管理药学研发实验记录,保证研发实验记录、数据的真实、准确、完整、可追溯,才能通过药学研制现场核查,确保产品获得注册批准。
药学研发实验记录的管理包括设计、书写、审核、保管、借阅等流程,记录的审核是其中重要的环节和步骤,记录审核可以及时的发现问题、改进问题,通过总结、分析以提高记录审核的效率,并反过来进一步促进记录的设计、记录的规范书写,本次就药学研发实验记录的审核要点进行总结。
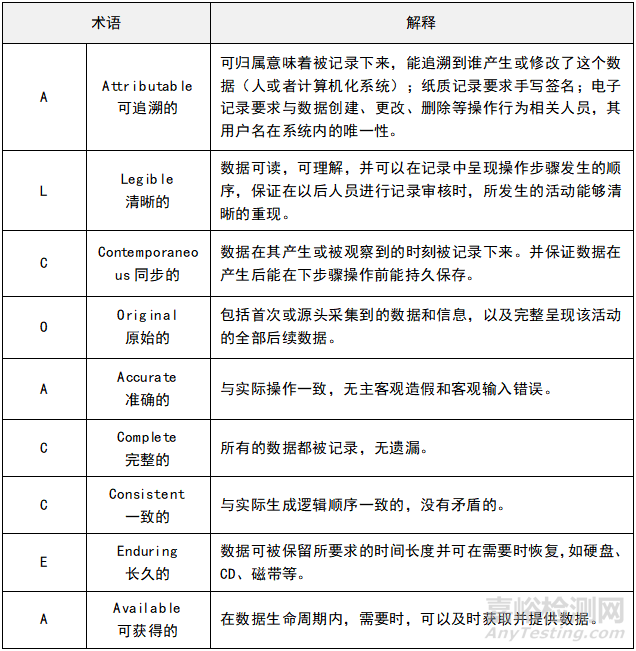
一、要熟悉数据可靠性(ALCOA+)的原则,适用于药品数据全生命周期的管理。
二、要组织各个部门建立实验记录的通用模板或专属模板,便于记录格式的统一、书写和审核。实验记录的内容通常应包括实验名称、实验目的、实验设计或方案、实验时间、实验材料、实验方法、实验过程、观察指标、实验结果和结果分析等内容,具体内容可根据研究的内容来确定。
三、应明确记录审核的责任,通过班组、部门、QA的层层审核,确保记录符合数据完整性的原则。
四、建立通用的审核要点,便于各人员、各部门进行审核,提高审核效率。

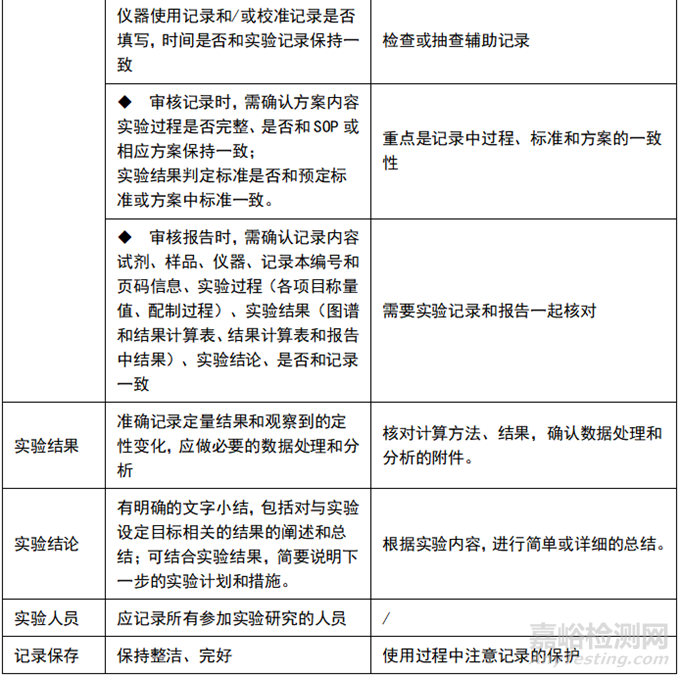
五、在审核、抽查记录过程中应注意及时总结,对共性问题进行培训教育,提高人员记录书写的规范性,提高对记录的认识,也可提高记录审核的效率。共性问题包括但不限于:实验信息不完整、记录不及时或漏记、修改不规范、实验人员/复核人员未签名签日期等,在培训时,要结合实际案例进行,会起到更好的培训效果。
综上,原始记录是进行了相应研制工作的证据性文件,也是申报资料的依据,通过记录审核,查漏补缺,保证记录和数据的真实、准确、完整、可追溯,确保研发项目通过核查、获得批准,所以研发人员要重视记录审核,规范记录审核。
参考资料:
GMP指南 质量管理体系2023版
《药品研究实验记录暂行规定》2000年
《药品记录与数据管理要求(试行)》2020年第74号公告
《药品注册核查要点与判定原则(药学研制和生产现场)(试行)》2021年第30号通告