医疗器械注册是指医疗器械注册申请人依照法定程序和要求提出医疗器械注册申请,药品监督管理部门依据法律法规,基于科学认知,进行安全性、有效性和质量可控性等审查,决定是否同意其申请的活动。技术审评作为注册管理的一个重要环节,审的是产品的安全性和有效性,是对与安全有效相关的证据进行系统评价的过程。
对于首次注册的产品,技术审评通常以《医疗器械安全和性能的基本原则》(以下简称《基本原则》)和《医疗器械 风险管理对医疗器的应用》为抓手开展,上述文件在对产品安全有效性进行系统评价的过程中有着非常关键的作用。
1、 基本概念
1.1 安全
按照《医疗器械风险管理对医疗器械的应用》(GB/T42062)3.26定义,安全是免除了不可接受的风险的状态。医疗器械是没有绝对安全,所谓的安全是相对风险而言。
1.2 有效
有效在字典的定义是“能实现预期用途”,医疗器械预期都是解决某个医疗问题。例如,高频手术设备的预期用途为在外科手术中,用于软组织的切割和凝血。那么实现软组织切割和凝血的能力即为高频手术设备的有效性。
评价一个产品是否能够上市是受益和风险的权衡,也是安全性和有效性的综合考虑。正如前文所述医疗器械没有绝对的安全,所谓的安全是相对风险而言,技术审评最终得出的结论是基于医疗器的有效性(受益)与医疗器械的安全性(风险)综合之后是否能够得出受益大于风险的结论。随着认知程度、教育医疗器梳质量-程度、社会经济、患者健康状态以及其他因素的变化,对安全有效的认知和判断可能也会不断的变化和发展。
1.3 系统性评价
系统性评价是对产品安全有效性进行深度和广度的综合评价。举例来说,有源医疗器械需要递交YY9706.102-2021电磁兼容检测报告,技术审评需要针对报告内容进行审评。但这并不是完整的深度评价过程。完整的深度评价过程应参考《医疗器械安全和性能基本原则符合性技术指南》,确定产品是否适用《医疗器械安全和性能基本原则清单》中A5.2c)h)A7.5、A7.6的相关要求以及证明符合该要求的方法,最后对符合性的证据文件进行评价见图1。
而电磁兼容性只是医疗器械产品应该考虑的单一风险,开展完整深度评价的同时还应对产品安全有效性进行广度评价,例如还应考虑产品的电气安全方面的风险、生物学方面的风险、能量相关的风险以及产品的有效性等。因此,认为技术审评就是审标准的符合性是有些片面的。符合标准只是证明产品在某一方面达到了行业共识,完成标准符合性的审评只是系统评价象限中的一个点或者局部面。对于技术审评来说,完成系统性评价是需要多年经验积累,而基本原则》可提供系统性评价的工具
2、医疗器械安全和性能的基本原则
2.1 背景
2005年,国际医疗器监管机构论坛(IMDRF)的前身CHTF(全球协调工作组)发布了第一版《基本原则》。《基本原则》发布后在全球范围内广泛运用、历经2012、2018年两次修订、完善,形成现行版本为构建科学的医疗器械监管体系,加快与国际接轨,国家药监局组织相关部门等同转化IMDRF2018版基本原则》,于2020年3月发布。
《医疗器械注册与备案管理办法》第十三条规定:医疗器械注册备案应当遵守相关法律、法规、规章、强制性标准,遵循医疗器安全和性能基本原则,参照相关技术指导原则,证明注册、备案的医疗器械安全、有效、质量可控,保证全过程信息真实谁确完整和可追溯。同时,医疗器械安全和性能基本原则清单也作为非临床资料列人注册申报资料要求。
2.2 框架及内容
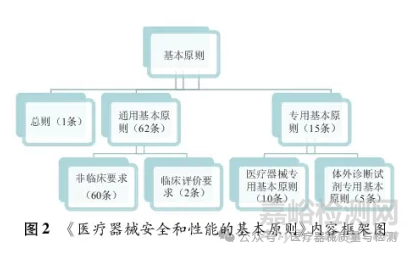
《基本原则》全文共78条,8016字,78条要求分为三个部分。
第一部分总则,强调了从事医疗器械行业的注册人/备案人所应具备的基本能力,即确定医疗器械安全和性能要求时应考虑全生命周期下的所有情况,医疗器械设计和生产不能脱离质量管理体系控制,同时注册人/备案人应当提供证明产品安全有效的证据第二部分是所有医疗器械适用的基本原则,明确了非体外诊断医疗器械和体外诊断医疗器械在设计生产过程中应关注的风险点,以及相关的性能要求第三部分按照非体外诊断医疗器械和体外诊断医疗器械两大类别,对特定医疗器械,分别补充了它们所应满足的安全和性能基本原则。如可吸收医疗器械、医学影像医疗器械、植人式医疗器、含药物成分的组合产品等。见图2.
2.3 技术审评过程中的作用
医疗器械安全和性能基本原则清单作为注册申报资料要求的一部分,整合了与安全有效性相关的基本要求,并纳人了符合性方法和客观证据,形成了一个从提出要求到证明要求的符合性闭环,构建了系统性评价的基础。技术审评就是以《基本原则》要求为索引,将注册申报资料中各项证据紧密联系起来,从安全性能基本要求出发对产品进行系统性评价,从而确保不遗漏任何重大风险点。这样的评价过程有助于提升医疗器械的安全性和有效性,保障患者的利益。如果《基本原则》中适用的条款完整,证明符合性所采用的方法合理、恰当,所提交证据全面、充分,则可认为产品符合安全有效基本要求,通过技术审评,获准注册。
3、医疗器械风险管理
医疗器械的安全性和风险之间紧密联系。《医疗器械风险管理对医疗器械的应用》(GB/T420622022)旨在帮助医疗器械制造商识别与医疗器械相关的风险,估计和评价相关的风险,控制这些风险并监视控制的有效性。《基本原则》清单的内容是注册申请人在设计和生产过程中,按照风险控制原则,对所形成文件进行逻辑性梳理。清单中安全性相关的要求,是各制造商在风险管理过程中识别出通用安全特征的总结和提炼,清单中的方法和客观证据则是用于证明制造商风险控制措施的有效性,最终的审评结论则是基于产品的剩余风险和产品有效性的综合考量得出的。因此掌握GB/T42062的风险管理思路在审评过程中结合制造商提供的风险管理资料,对于完成产品安全有效性系统性评价至关重要。《基本原则》是适用于所有医疗器械的通用要求,已识别出的风险包括生物相容性、微生物污染、辐射安全、环境和使用条件、电气安全、电磁兼容、有源医疗器械的连接等方面的风险,并不能够保证穷举所有产品的风险,风险管理的思路还有助于提出更多安全性相关的问题以查漏补缺。
4、变更注册审评思路
《医疗器械注册与备案管理办法》第八十条规定,对于变更注册申请,技术审评机构应当重点针对变化部分进行审评,对变化后产品是否安全、有效、质量可控形成审评意见。技术审评应以《基本原则》和医疗器械风险管理对医疗器的应用》为指导,关注变化部分对产品安全有效性的影响。如发现有任何影响,注册人需提供充分的证据来证明变化后产品的安全有效性。变化部分对产品安全有效性的影响可能是多方面的,应综合考虑。例如产品技术要求中载明的与人体接触部分的材料发生变化,就需要根据产品适用范围和技术特征,全面考虑变化部分对产品技术要求中的性能指标、生物学特性、清洁消毒灭菌稳定性、临床有效性等方面的影响,并提供相应的注册申报资料。
5、结语
通过合理运用《医疗器械安全和性能的基本原则》和《医疗器械风险管理对医疗器的应用》,所有的审评意见都将有依据可循。如果注册申请人能够提供完整且充分的医疗器械安全和性能基本原则清单以及风险管理资料,将有利于提高技术审评的效率和质量。同时,技术审评的一致性、规范性、科学性和系统性也将得到大大提升。