您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-04-29 08:14
放射性治疗药物(Therapeutic radiopharmaceuticals)是将具有细胞毒性的放射性核素选择性地输送到病变部位,利用放射性核素的衰变特性释放射线或粒子,对病变细胞产生杀伤作用,从而达到治疗目的的一类药物。通常由放射性核素和非放射性部分组成。放射性核素是指能自发衰变并释放粒子或射线的不稳定核素。非放射性部分是指与放射性核素结合并将其递送至体内靶标部位的配体或载体,如多肽、蛋白、脂质体和小分子。简单示意图如下:

根据给药途径不同可分为系统给药和局部给药,系统给药包括口服或静脉给予的同位素药物(例如碘[131I]化钠、氯化镭[223Ra]等),和放射性配体药物(例如lutetium Lu 177 dotatate、lutetium Lu 177 vipivotide tetraxetan等);局部给药包括植入放射性粒子(例如碘[125I]密封籽源)和放射性栓塞微球(例如钇[90Y]微球)等。
关于放射性治疗药物的非临床研究,虽然特定针对该品类的指导原则还不多,但随着这类药物研究的增加,国内外监管机构陆续颁布了一些,罗列如下:
2011, FDA, Nonclinical-Evaluation-of-Late-Radiation-Toxicity-of-Therapeutic-Radiopharmaceuticals.
2018, EMA, Draft-guideline-non-clinical-requirements-radiopharmaceuticals.
2018, FDA, Microdose-Radiopharmaceutical-Diagnostic-Drugs--Nonclinical-Study-Recommendations.(诊断)
2019, FDA, Oncology therapeutic radiopharmaceuticals nonclinical studies and labeling recommendations guidance for industry.
2023, IAEA, Guidance for preclinical studies with radiopharmaceuticals.
2024, NMPA, 放射性治疗药物非临床研究技术指导原则。
结合已颁布的各个法规和一些案例,分享下放射性治疗药物的临床前研究策略。
与传统药物开发路径类似,放射性治疗药物早期也会先进行体外研究包括理化表征、细胞吸收、亲和力、特异性、体外代谢、血清稳定性等,之后进行体内研究,并逐步确认候选分子,进入临床阶段。当然,放射性治疗药物也有其特别之处,从这类药物作用机理不难看出,其发挥药理学作用需要依赖某些组分将放射性核素靶向递送至特定组织(如肿瘤)的特定靶点。因此,药物在靶组织的蓄积、滞留时间、放射活性就比较重要,可以通过体内组织分布考察。另外,靶向递送分子的特异性决定了是否有脱靶毒性。而且,靶向递送分子有些是多肽或小分子,还要按照化药思路,考察体外肝细胞代谢,以评估种属差异。放射性核药开发过程示意图如下。
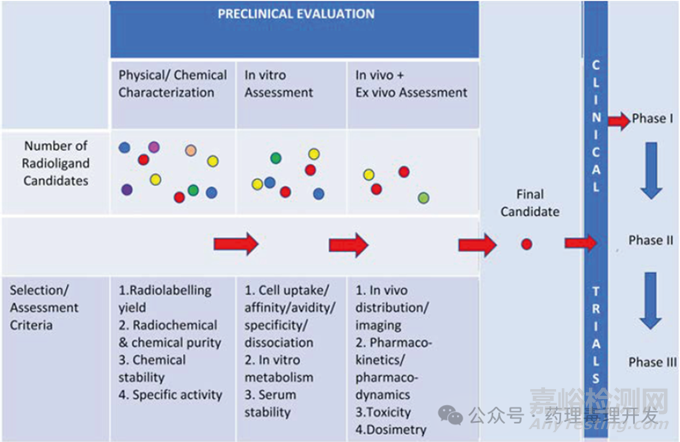
主要药效学
1、法规要求
CDE:在首次人体临床试验之前,应当根据作用靶点和作用机制等选择合适的模型,开展主要药效学试验。例如抗肿瘤药物应研究抗肿瘤活性、肿瘤组织摄取等。作用机制研究应评估药物与靶点的亲和性(如结合、解离能力等)、选择性、特异性等。此外,评估药物对其他受体、酶、离子通道、转运体等的影响,分析可能的脱靶作用,有助于评估药物的选择性和特异性。
EMA:建议围绕靶点/受体特征开展相关研究。应证明非放射性部分是否具备药理学活性。
FDA:在肿瘤治疗放射性核药非临床研究指南中提出,主要药效学部分应该在首次人体临床试验之前完成概念验证,证明药物在肿瘤部位的吸收和抗肿瘤活性。可以通过体外实验研究药物的作用机制,比如体外的靶点结合和抗肿瘤活性。体内外研究均需要选择合适的评价终点。
2、案例:
以2010年以后批准的3个核药产品作为案例,分别是2013年上市的拜耳的Xofigo,又名223Ra dichloride,用于治疗晚期骨转移型去势抵抗性前列腺癌;2018年上市的Advanced Accelerator Applications公司的Lutathera,又名177Lu dotatate,用于治疗生长抑素受体阳性的胃肠道及胰腺的神经内分泌性肿瘤(GEP-NETs);2022年上市的Novartis的Pluvicto,又名177Lu vipivotide tetraxetan,用于治疗前列腺特异性膜抗原(PSMA)阳性、既往接受过治疗的转移性去势抵抗性前列腺癌患者。
2.1 Xofigo
Xofigo的结构比较简单,就是一个放射性镭元素连接两个氯离子。Xofigo的作用机理是镭进入人体会被当做钙质吸收,大量聚集在骨骼内,通过镭223释放的α射线粒子使邻近细胞的DNA双键断裂,达到杀伤的目的。这就不难理解为什么Xofigo适应症是癌症骨转移了。有点类似于碘131,一种亲甲状腺的物质。镭223属于亲骨物质。这类核素不需要靶向载体递送。
体外药理研究内容:1)5项研究评估α射线粒子对NHIK3025、A549细胞的DNA损伤作用、存活、细胞周期影响等;2)对人、小鼠破骨细胞分化、发育的影响。
体内药理研究内容:1)在乳腺癌骨转移模型中的剂量探索研究;2)在乳腺癌骨转移模型中的作用机理研究、动物生存研究。
Xofigo不是通过偶联多肽或者抗体实现靶向,因此未开展非放射性部分的靶点亲和力、选择性、特异性等研究。主要围绕Xofigo的放射性部分的杀伤机制、活性、亲骨性及具备一定临床转化性的动物药效进行了评估。
2.2 Lutathera
Lutathera是生长激素抑制素类似物的放射性标记药物。生长激素抑制素受体2(Somatostatin receptor 2, SSTR2)在神经内分泌瘤中过表达,Lutathera结构中的生长激素抑制素类似物可以将放射性元素177Lu靶向递送至肿瘤部位,并发挥肿瘤杀伤作用。
体外药理研究内容:1)竞争性取代Somatostatin与细胞表面的SSTR1、2、3、4、5不同受体的结合(提交的文献资料);2)竞争结合CA20948胰腺癌细胞somatostatin受体;3)在大鼠AR42J胰腺癌细胞中的内吞研究。
体内药理研究内容:CA20948大鼠胰腺癌移植瘤模型中的组织分布及药效研究(单次、多次,IV给药)。该研究还伴随非放射冷药阻断研究。
Lutathera的主要药效学研究均是采用热药开展的,研究内容相对规矩,体外探索结合特异性,既包括人为构建细胞,也包括了目标肿瘤细胞。体内考察了在目标瘤种中的药效情况,既包括单次给药,又涵盖多次给药,还通过给予冷药阻断热药的作用确证了发挥作用的物质基础。将组织分布整合进药效试验,充分利用了动物资源。
2.3 Pluvicto
PSMA在前列腺癌细胞表面过表达,Pluvicto通过靶向PSMA的多肽配体,将177Lu递送至肿瘤部位,并形成杀伤,如下图所示。

体外药理:1)177Lu-PSMA-617与PSMA阳性前列腺癌细胞系LNCaP的结合研究;2)PSMA-617阻断177Lu-PSMA-617与LNCaP的结合研究;3)提交文献资料,内容包括非标PSMA-617与重组人PSMA蛋白的分子亲和力、细胞亲和力和竞争实验;4)177Lu-PSMA-617在PSMA阳性和阴性PC-3人前列腺癌细胞系中的内吞作用研究;5)177Lu-PSMA-617对PSMA阳性和阴性PC-3细胞活力、生存的影响。
体内药理:1)177Lu-PSMA-617对RM1-hPSMA小鼠同源移植瘤模型的药效学研究;2)177Lu-PSMA-617在PSMA阳性和阴性PC-3人前列腺癌细胞小鼠移植瘤模型中的组织分布、药效学研究。
次药药效学:175Lu-PSMA-617非放射性药物与不同受体、离子通道、酶和转运体的相互作用研究。另外,证明了非标、冷标供试品无药理学活性,即杀伤作用是依赖177Lu实现的。
Pluvicto是通过结合PSMA发挥作用,因此药效学部分重点研究了177Lu-PSMA-617与表达PSMA的细胞的结合、阻断、内吞及活性。种属选择性方面虽然未提供数据,但体内药效采用的是人PSMA转基因的鼠RM-1细胞,说明非放射性多肽部分PSMA-617可能不与小鼠PSMA结合。另外,Pluvicto开展了比较全面的脱靶风险研究,类似化药的开发思路。
安全药理学
1、法规要求
CDE:通常在首次人体临床试验之前完成安全药理学试验。
EMA:未提及。
FDA:对于治疗肿瘤的产品,单独开展针对心血管、呼吸和神经系统安全药理学研究并不是必须的,可以伴随毒理研究或组织分布研究开展。
2、案例
Xofigo开展了223镭对大鼠神经系统、呼吸系统,比格犬心血管系统安全药理学研究。
Lutathera开展了175Lu冷标化合物对大鼠神经系统、呼吸系统,比格犬心血管系统安全药理学研究。另外,开展了体外hERG试验。
Pluvicto开展了非标PSMA-617和冷标175Lu- PSMA-617化合物(1:1混合)对大鼠神经系统、呼吸系统,小型猪心血管系统安全药理学研究。另外,开展了体外hERG试验。
从以上可以看出,虽然FDA支持安全药理学研究伴随毒理或组织分布实验中开展,但3个已上市产品均采用了单独研究的形式。
药代动力学
1、法规要求
CDE:特别强调,放射性治疗药物的组织分布试验,是支持临床试验中人体组织分布、器官辐射剂量估算和安全性评估的关键试验,应在首次人体临床试验前完成。吸收试验可以伴随组织分布开展。组织分布实验其它考量点包括单种属、双性别、多剂量、采样不小于5个半衰期,鼓励在模型动物中开展。其它药代研究内容包括体内、外代谢研究,排泄研究。如果载体或配体是新结构化合物,应提供该部分的ADME信息。
EMA:应开展生物分布、消除研究,为毒性靶器官鉴定,临床组织、全身放射性剂量估算提供依据。
FDA:应在IND之前开展动物生物分布研究以指导临床生物分布试验的剂量选择。可以单种属、双性别(除非适应症为单性别)设计,采样周期至少覆盖5个半衰期。冷标和热标化合物比例应与临床拟用产品相当。鼓励进行多剂量水平设计。排泄数据可以从尿液和粪便中收集。
在药代动力学法规层面,CDE和FDA的要求是比较类似的。当然,指南中内容还包括建议采集的组织、临床放射剂量换算等,篇幅所限,只能提纲挈领。
2、案例
Xofigo开展了:1)223镭在BALB/CA雌性小鼠单次给药PK和生物分布试验;2)223镭在犬中单次给药生物分布和单次给药毒性试验;3)223镭在犬中重复给药长期放射毒性试验;4)223镭在小鼠尿和粪便中的排泄研究;5)唑来膦酸治疗时,223镭在小鼠体内的PK和生物分布研究。
Lutathera开展了:1)177Lu-DOTA-Try3-Octreotate在CA20948胰腺癌移植大鼠中的生物分布研究;2)177Lu-DOTA-Try3-Octreotate在AR42J胰腺癌移植大鼠中的生物分布研究(添加非标化合物Try3-Octreotate进行阻断);3)冷标化合物175Lu-DOTA-Try3-Octreotate在大鼠、犬和人肝细胞、肾匀浆物体外代谢研究;4)冷标化合物175Lu-DOTA-Try3-Octreotate体外CYP450酶抑制、诱导实验;5)177Lu-DOTA-Try3-Octreotate在CA20948胰腺癌移植大鼠中的排泄研究;6)DDI方面研究了对BCRP、OATP1B1、OATP1B3、OAT1、OAT3、OCT2、OCT1的抑制研究,并通过Caco-2评价了P-gp介导的相关作用;7)多次给药PK数据源自重复给药TK;8)其它PK相关研究还包括体内外大鼠血浆稳定性、血浆蛋白结合实验等。
Pluvicto开展了:1)冷标化合物175Lu- PSMA-617、非标化合物PSMA-617与大鼠、小型猪和人血浆蛋白结合、血浆稳定性实验;2)177Lu- PSMA-617小鼠组织分布研究(提交的文献资料);3)177Lu- PSMA-617雄性大鼠单次IV给药组织分布研究;4)排泄研究伴随在组织分布试验中开展;5)冷标化合物175Lu- PSMA-617、非标化合物PSMA-617大鼠单次给药TK研究(伴随毒理研究中考察);6)冷标化合物175Lu- PSMA-617、非标化合物PSMA-617小型猪单次给药TK研究(伴随毒理研究中考察);7)175Lu- PSMA-617、PSMA-617体外大鼠、小型猪和人肝脏、肾脏代谢稳定性研究。
毒理学
1、法规
CDE:评价分为辐射相关毒性和非放射性部分的毒性。如果药效学和组织分布试验剂量能够覆盖毒理学剂量,放射性治疗药物的一般毒理学试验可以伴随在这些研究中开展,但动物数量、评价指标应符合毒理研究相关指南要求。如果放射性核素是全新的,建议啮齿+非啮齿双种属开展一般毒理试验。如果核素已有人用经验,且临床风险可控,可选择一种相关动物种属开展一般毒理研究。除放射性药物即所谓的“热药”毒理研究外,需开展“冷药”的一般毒理试验,原则上采用啮齿+非啮齿双种属。
以上是支持IND毒理研究要求。支持后续临床试验和上市时,还需要开展更长周期毒理研究和迟发放射性毒性评估。更长周期毒性研究要求可参考ICH M3(R2)、S6(R1)和S9相关要求。不过,如果当临床给药次数有限(如给药2次或3次)、配体或载体仅用于递送目的且给药剂量低(如在微克范围内)、配体或载体的半衰期短且给药频率低时(如每4至8周给药一次),可能不需要开展冷药的更长周期的一般毒理学试验。迟发放射性毒性评估可以在一个种属中开展,观察周期至少为1年,且应在Ⅱ期临床试验开始前完成。迟发放射性毒性至少设置4个剂量,以确定NOAEL和迟发放射性毒性的剂量相关性。该研究还需设置冷药对照组。选择的最高剂量应产生急性放射性毒性,该剂量应至少是人体最大拟用剂量或关键器官的辐射耐受剂量(TD5/5外照射)的2倍。
放射性治疗药物通常不需要开展遗传毒性、生殖毒性和致癌性研究。
放射性治疗药物当配体或载体为多肽、蛋白等大分子时,应开展免疫原性和免疫毒性研究。
放射性治疗药物应在首次临床试验之前,完成制剂安全性研究。
EMA:建议开展啮齿和非啮齿类动物的毒理研究。如果非放射性部分无药理活性(大多数核药属于这类情况),则可以在1个种属中开展,首选啮齿类动物。局部耐受性、安全药理研究可以伴随单次给药毒理试验开展。
如果非放射性部分不是生物技术产品,应开展遗传毒性研究,比如Ames。当然,如果适应症为肿瘤,则不用开展。
生殖毒性试验和致癌性试验通常不开展。
GLP依从性方面,由于放射性核药的特殊性,有些毒理研究结果可能来自non-GLP条件,比较常见的是组织分布研究中伴随的毒理学评价。因此,通常会开展GLP条件下的冷标化合物毒理学研究,以弥补可能的风险。
FDA:对于肿瘤适应症放射性核药,应开展放射性毒性和配体相关的毒性。可以单独开展,也可以伴随组织分布试验开展。如果供试品是纯核素(如无配体偶联),则不用开展毒理研究。关于放射性相关毒性评估,FDA认为不用开展放射性药物的一般毒理研究,动物生物分布研究中伴随毒理学终点考察即可。另外,最好提交放射性核素相关的器官特异性毒性的文献资料作为辅助性证据。关于配体介导的毒性,应开展冷药的一个种属的一般毒理学研究(这点与CDE的要求不同)。
以上是支持IND的毒理试验要求,对于支持后续临床试验和产品上市的毒理研究还涉及更长周期的毒理试验。其中,涉及冷药部分的配体介导的更长周期毒性要求与CDE一样,两个机构的关于这部分的描述基本是中英文翻译版。另外,FDA提到迟发性放射毒性可以和更长周期一般毒理研究合并开展。这点没太明白,指的是采用热药在non-GLP条件下开展的迟发性放射毒性/更长周期毒理合并研究可以接受?毕竟EMA提到,来自组织分布的non-GLP放射毒性研究结果是可以接受的。当然,如果GLP实验室本身就具备资质,能开展热药的毒理研究那是最好不过了。
FDA不建议开展遗传、生殖和致癌性试验。
2、案例
2.1 Xofigo
Xofigo开展了:1)223镭小鼠单次给药毒性试验;2)223镭大鼠单次给药毒性试验;3)223镭在犬中单次给药生物分布和单次给药毒性试验;4)大鼠每4周给药一次重复给药223镭4次,恢复期1年毒理研究(合并考察了迟发性放射毒性)。另外该研究还设置了4组单次给药组别。5)大鼠重复给药223镭12个月恢复期4周(3个剂量,每4周给药一次,共12次)毒性试验;6)犬重复给药223镭20周恢复期4周毒性试验(每4周一次,共6次);7)兔局部刺激性试验。
Xofigo结构中涉及冷药部分,开展的均是热药的毒理研究。Xofigo是首个上市的223镭核素,开展的啮齿+非啮齿多种属单次和多次给药毒性研究。当然,单次毒理试验可以与组织分布试验合并开展。Xofigo临床给药方案是每4周一次,共给药6次。非临床重复给药周期也是支持的。
2.2 Lutathera
Lutathera开展了:1)冷标化合物在大鼠、犬中单次给药毒性试验;2)冷标化合物在大鼠中42天重复给药毒性试验(3个剂量,每2周给药一次);3)冷标化合物在犬中42天重复给药毒性试验(3个剂量,每2周给药一次);4)遗传毒性:冷标化合物Ames试验、L5187Y TK+/-小鼠淋巴瘤细胞试验。
Lutathera临床每8周(±1周)给药一次,共给药4次。非临床毒理研究均采用冷标化合物开展。体内药效中有伴随热药的部分毒理学指标考察。
2.3 Pluvicto
Pluvicto开展了:1)冷标化合物175Lu-PSMA-617和非标化合物PSMA-617按1:1混合后,大鼠单次给药毒理研究;2)冷标化合物175Lu-PSMA-617和非标化合物PSMA-617按1:1混合后,小型猪单次给药毒理研究;3)非标化合物PSMA-617雄性大鼠重复给药毒性研究(每周1次,给药4次);4)非标化合物PSMA-617的Ames试验。未开展生殖毒性和致癌性试验。
Pluvicto临床给药方案为每6周一次,给药6次。从非临床研究内容看,似乎未披露太多热标药物的毒理研究数据。因临床目标人群为男性,重复给药毒性仅在雄性动物中开展。

来源:药理毒理开发


