您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-05-11 08:32
摘要
1995年,生物药剂学分类系统(Biopharmaceutics Classification System,BCS)的引入,为描述药物的生物药剂学行为提供了一种简单的方法。溶解度和渗透性是其主要参数之一,它们决定了药物吸收的量,也决定了药物的生物利用度。本综述的目的是总结溶解度和溶出度介质的演变,以及如何将其用于现代药物开发。多年来,为更好地预测药物的体内溶解度和溶出度,引入了生理相关介质和缓冲液。对水、缓冲液、药典介质、胶束增溶介质和生理相关介质进行了综述,目前,仍无通用的介质评估每种药物的溶解度或药品的体内溶出行为。为优化体内的可预测性,对介质进行了许多改进;例如,目前缓冲液中的磷酸盐浓度似乎太高,无法关联体内的碳酸盐缓冲液浓度。改进后的介质与人类肠道液体的组成更相似。BCS作为一种风险管理工具被引入监管科学,在特定条件下可用于豁免生物等效性研究。目前不同的指南对剂量-溶解度比的定义不同。如阿莫西林那样,可能会给全球运营公司带来更多的困惑。所以,统一BCS指南是非常必要的。
关键字:生物药剂学分类;溶出度;体内外相关性;溶解度。
引言
1995年,生物药剂学药物分类系统(BCS)的引入,为描述药物的生物药剂学行为提供了一种简单的方法。溶解性和渗透性是其主要参数之一,它们决定了药物吸收的剂量,从而决定了其生物利用度。此处溶解度是指水溶性。监管指南根据活性成分(API)在水中的溶解度对其进行分类。然而,人体胃肠道的生理结构很复杂,会对药物的体内溶解度产生很大影响。在过去的几十年里,为了更好地预测药物的体内溶解度,引入了生理相关介质和缓冲液。同样,溶出度方法最初是作为质量控制方法开发的,与体内的相关性不高,有时甚至没有相关性。然而,现今的监管机构希望看到更多的体内相关的溶出度规范和有区分力的溶出度方法,可以检测到药物的关键质量属性的变化。只有利用BCS的基本原理以及体内相关检测和预测才能以质量源于设计的方式开发产品。这突出了BCS监管分类和基于预测API体内行为/体内溶解所需的BCS原理的科学机制信息之间的差距。
本综述的目的是总结溶解度和溶出度介质的演变,以及如何将其用于现代药物开发。其文献检索是在2014年9月使用四个数据库进行的,分别为ISI Web of Science、SciFinder、Scopus和Google Scholar。经过仔细筛选,除了科学期刊上发表的原始研究外,还有报告、学位论文和会议投稿,检索时没有日期或语言限制。随后在2016年7月进行了检索更新。关键词如下:BCS、生理相关介质、溶解度、空腹状态、进食状态、药物溶解性、固有溶解性、模拟肠液、模拟胃液、缓冲溶解介质、表观溶解度和溶解速率。本综述共纳入51项研究,其中一些同时使用了摇瓶法和溶出度测试法,而46项仅使用摇瓶法,20项仅使用了溶出度测试法。
将每种介质组成及药物溶解度数据制成表格。介质组成的相关信息摘自官方指南:欧洲药品管理局(EMA)、食品和药物管理局(FDA)、加拿大卫生部、巴西卫生监督局(ANVISA)、世界卫生组织(WHO)以及巴西、欧洲和美国药典。
水中的溶解度
从生物药剂学角度,水溶性是药物的一个重要的理化性质。溶解度是指在恒定温度和压力下溶解到溶剂中以达到饱和溶液的药物的量。溶解度以溶解在一定体积或质量的溶剂中的溶质的最大体积或质量表示(1)。
Higuchi和Connors(2)提出的摇瓶法目前被广泛用于药物溶解度测定。它测定了药物达到平衡后的热力学溶解度。在特定温度下将过量的药物溶解到特定的介质中。根据溶解速率和搅拌类型,可在数小时或数几天达到平衡。平衡阶段,药物和饱和溶液可通过过滤或离心分离(3)。
美国药典(USP)用更通俗的术语来描述溶解度,并将其定义为溶解1份重量固体或1份体积液体所需的溶剂的体积份数(4),如补充材料1中所述。
USP所描述的水溶性通常指室温下水中的参考溶解度。大多情况下,介质的pH和成分会影响药物溶解度。如pH依赖性官能团可被电离,对溶解度影响很大(5)。
因此,水中溶解度并不代表胃肠道溶解度,特别是对于亲脂性和难溶性的药物。
水
文献表明,不同等级的水用作测定药物溶解度的介质:双蒸馏水(6-8);双去离子水(9),Milli-Q®纯化水(10),18.2MΩ.cm/ 0. 22μm(11);Milli-Q®超纯水,电导率小于0.1μS c m(12,13);超纯水Elix® Millipore(14);蒸馏水(5,15-21)去离子水(22-26);蒸馏、去离子和过滤水(27);纯净水(28-36);去矿物质水(37)。51项研究中19项(37%)未规定水的类型(38-56)。
如果用于质量控制分析的分析级水没有具体的药典要求,它仍须满足USP纯净水定义的要求。USP在其总章1231(4)中列出了以下水:蒸馏水、新鲜蒸馏水、去离子水、新鲜去离子水、去离子蒸馏水、过滤水、高纯水、无氨水、无二氧化碳水、无氨和二氧化碳水、脱气水、新近煮沸的水、无氧水、细菌内毒素检测用水、无有机水、无铅水、无氯水和热水。这些水中的每一种都具有特定检测所需的特定属性。如脱气水不影响药品的溶解度,但改变药品的溶出率。在USP性能验证测试时,如果溶出介质未充分脱气,气泡会附着在泼尼松片的粉末和原料颗粒上,使其漂浮在介质中,桨下发生的堆积较少,这增加了溶出率,测试可能不符合溶出规范。
缓冲液
该文献综述发现,32.6%将pH5.8-8.0的磷酸盐缓冲液作为摇瓶法的介质(46项研究中的15项)(8,11,19,31,32,37,41-43,45,46,48,51,57,58),40.0%将其作为溶出介质(20项研究中的8项)(10,11,22,24,51-53,56)。醋酸盐缓冲液的使用频率较低,46项研究中有4项(8.7%)(41,42,45,49)将其作为摇瓶法的介质,20项研究中有2项(10.0%)将其作为溶出介质(52,56)。磷酸盐和醋酸盐缓冲液称与生理液体有相似的渗透压和离子强度,是测定溶解度的常用缓冲液(磷酸盐缓冲液为27.3%,醋酸盐缓冲液为9.1%)。
药物的盐溶解度不仅取决于药物的浓度,还取决于反离子的浓度。缓冲液可以引入反离子,由于络合物的溶解度较低,药物可能会与之沉淀。另一方面,由于盐析效应,高浓度的反离子会降低盐的溶解度。如Kambayashi所报道的,在中性或碱性溶液中,带正电的碱性药物会因pH的变化而以磷酸盐的形式沉淀。Bergström报告了一组25种药物的相似溶解度变化,并描述了在整个pH范围内观察到的溶解度的变化很大。物质的固有溶解度与不带电和完全带电的药物种类和磷酸根离子有关(60)。Völgyi报道了盐酸二丙诺啡、盐酸可待因和磷酸盐,以及盐酸利多卡因和磷酸盐的盐溶度。在这项研究中,当用盐酸调节pH以降低缓冲液的pH时,除磷酸盐离子,氯离子也引起了溶解度的变化(61)。
在这种条件下,药物,特别是含有表面活性的药物,可以形成二聚体、三聚体或更高级低聚物形式的胶束或自结合聚集体(62)。许多非甾体抗炎药,如消炎痛、双氯芬酸、布洛芬、酮洛芬和萘普生,往往通过形成混合带电胶束或胶束样结构而自结合(63)。在这样的系统中,溶解度-pH曲线不能用Henderson-Hasselbalch方程(62)准确描述。Avdeef用pDISOL-X程序对Higuchi 在1953年(64)发表的数据进行重新评估发现,药物分子的二聚体会导致Henderson-Hasselbalch方程的偏差。该方法是基于Volgyi发表的关于溶解度的预估,它与Henderson-Hasselbalch方程无关(19)。
本报告中,43项研究只有1项使用了改良Hank‘s缓冲液(10,48),是以碳酸氢盐为基础的缓冲液(表I),用于摇瓶法。Krebs缓冲液,一种以碳酸氢盐为基础的缓冲液,用于测定布洛芬的药物溶解度(48),并作为美沙拉嗪包衣片的溶出介质(65)。该缓冲液接近生理pH和渗透压,常用于肠道吸收研究。Krebs缓冲液缺点是对储存条件要求较高。需用5%的二氧化碳持续通气,并在顶部加液体石蜡和/或用完全密封的装置使缓冲液稳定(65)。
表I.溶解度试验中使用的磷酸盐缓冲液(PB)和Hank缓冲液(改性碳酸氢盐缓冲液)的组成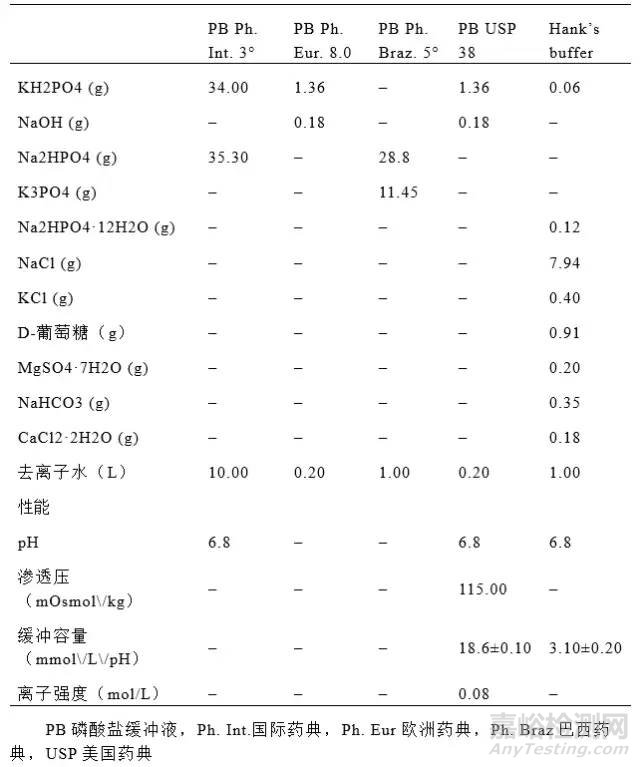
在pH 4.0-6.8范围内使用柠檬酸盐/磷酸盐缓冲体系的McIlvaine缓冲溶液(6.5%),46项研究中有3项用于摇瓶法(5,27,48),20项研究中有2项作为溶出介质(10.0%)(5,27)。选择马来酸盐(32)、Britton-Robinson通用型(19,25,39)和甘氨酸缓冲液(31)用于摇瓶法的比例分别为2.2%(46项研究中的1项)、6.5%(46项研究中的3项)和2.2%(46项研究中的1项)。
很明显,溶解度测定有很多不同的缓冲液可选择。是否可以将其中一种作为通用的首选介质需要研究证明。磷酸盐缓冲液是目前最常用的介质,但其溶解能力不足,无法预测难溶性药物的体内溶解度(66,67)。
USP药典介质
根据FDA(68),含胃蛋白酶的模拟胃液(USP-SGF)和无酶缓冲液SGF(SGF空白)或含胰蛋白酶的模拟肠液(USP-SIF)和无酶缓冲液(SIF空白)较其他缓冲液更能反映胃和小肠的生理状况。USP-SGF和USP-SIF的组成见补充材料2(4)。综述发现,17.4%(46项研究中的8项)(13,15,17,21,26,40,41,49)和10.0%(20项研究中的2项)(13,17)使用SGF与摇瓶法进行溶出试验,通常采用无酶缓冲液SGF进行。
Dressman等人(69)描述了空腹状态的模拟胃液,其含有牛黄胆酸钠和卵磷脂可更好地模拟人类胃液成分。
这些介质一般被称为生理相关介质,稍后将在消化道的肠段中讨论。
在模拟肠液中,将pH由7.5改为USP25中要求的6.8(70)。若使用的pH大于7.5,应提供科学的说明,因其超出了生理pH范围 (9)。有些USP各论的pH为8.0甚至更高(71)。双氯非那胺片、氯噻嗪片、依他尼片、甲基多巴和氯噻嗪片、利血平和氯噻嗪片以及熊去氧胆酸片,溶出介质pH为8.0;康复龙片和格列本脲(微粉)片,溶出介质pH为8.5;萘啶酸片,溶出介质的pH为8.6;格列本脲(非微粉)片,溶出介质的pH为9.5;而左旋甲状腺素钠片,溶出介质的pH达10(71)。8.7%(或46项研究中的4项)(15,17,26,49)用SIF空白为溶出介质,10.0%(20项研究中的2项)(17,24)用SIV空白为溶出介质。
比较SIF空白和标准的pH6.8磷酸盐缓冲液,如国际药典(72)(表 II和表III),其组成可能存在差异。然而,正如Stippler等人所示,这些缓冲液具有类似的性质,包括渗透压、离子强度和缓冲容量(72)。在布洛芬、甲硝唑和吲哚美辛速释固体口服制剂的溶出度试验中,溶出介质是可以互换的。研究认为,只有已知溶解度受阳离子影响的情况下,才需要关注这两种阳离子相互取代。这与Almukainzi等人报告的结果一致(73)。使用钠和钾的缓冲液和SIF缓冲液使纤维素硬胶囊的崩解时间不同,从而导致不同的体外溶出行为。此外,Ropers等人(74)报告了钠和钾反离子对阴离子表面活性剂如烷基和十二烷基硫酸盐的胶束形成的影响。据报道,反离子的结合以Na+>K+的顺序减少。因此,使用钠缓冲液而非钾缓冲液可避免表面活性剂因反离子相互作用而沉淀。
表II. 空腹和进食状态下模拟肠液的组成
FaSSIF空腹状态模拟肠液,FaSSIF-V2空腹状态模拟肠液(第二代),FeSSIF进食状态模拟肠液,FeSSIF-V2进食状态模拟第三版肠液(第二代)
表III.生理相关介质和含水缓冲液在药物溶解度方面的性能比较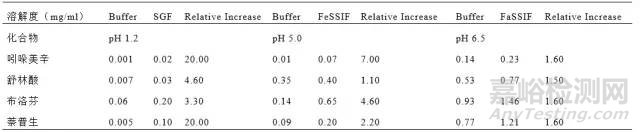
本报告显示,pH6.8的磷酸盐缓冲液是溶出研究中最常用的介质(35.0%)。含水缓冲液和药典介质模拟了胃或小肠中典型的pH、离子强度和渗透压。它们并不代表胃肠道(GI)生理条件的所有情况(如粘度、表面张力)(75),通常只能提供与体内数据的后验相关性(76)。此外,磷酸盐缓冲液不能在生物豁免中区分托拉塞米产品间的差异,根据BCS分类,托拉塞米是BCS I类药物。在这种情况下,pH5.0介质具有更好的区分力,优于FDA推荐的介质(75)。
最近,Krieg等人(77)比较了碳酸盐缓冲液和磷酸盐缓冲液。其结果将在碳酸氢盐缓冲液中详细讨论。他们指出,缓冲液的浓度是体内的相关性重要的因素。此外,上述介质不能模拟食物对药物释放的影响。因此,开发适用于空腹和进食状态的检测介质和方法来模拟特定的生理条件是显而易见的。
药物胶束增溶作用
胶束体系可以提高难溶性药物的溶解度并增加其生物利用度(78)。Zangenberg等人(79)开发了一个动态脂肪分解模型,其中甘油三酯的水解速度由加入的钙离子控制。这个模型模拟了进食状态脂类在体内消化和吸收过程。
他们观察到普罗布考和达那唑在脂解过程中的不同溶出曲线,并将其与亲脂性联系起来。普罗布考的log P超过10,达那唑的log P为4.5(79)。普罗布考在水中的溶解度取决于其在亲脂相和亲水相之间的分配。达那唑的溶出度取决于水的增溶能力。该模型提出并作为一种模拟进食状态下,研究脂解过程中药物从制剂中的溶出方法。
在最近的一项研究中,Ottaviani等人(80)研究了表面活性剂及其临界胶体浓度(CMC)对药物增溶关系。研究表明,在模拟空腹状态的肠液(FaSSIF)中,CMC比亲脂性(LogD)能更好的预测药物增溶情况。
Kaukonen等人(81)的比较了达那唑和其他四种模型药物在空白缓冲液和不同甘油三酯组成的胶束中的溶解度。研究了消化酶对难溶性药物(达那唑、灰黄霉素、地西泮、桂利嗪和卤泛曲林)的增溶作用。补充材料3中列出的数据表明,与纯胶束或缓冲液相比,当甘油三酯被水解时,由单、双和甘油三酯组成的混合胶束具有更高的增溶能力。据报道,其增溶能力灰黄霉素是10倍,地西泮是17倍,达那唑是178倍,桂利嗪是1600倍,而卤泛曲林是10720倍。
这种消化缓冲体系可用来模拟脂质与肠道环境对药物溶解度的影响。然而,该体系仍然不能完全说明影响肠道溶解的因素。
生理相关介质
在过去十年中,生理相关介质被广泛用于测定药物溶解度和作为溶出介质,尤其是对于BCS II类药物(14项研究或25.0%)(8,9,13,22,27,33,35,42,44,45,47,51,54,55)以及BCS I类(4项研究或7.1%)(33,34,49,51)、III类(2或3.6%)(33,51)和IV类药物(2或3.6%)(33,51)。如上所述,生理相关介质通常含有可能存在于人体胃肠道中的成分,例如胆盐和卵磷脂,旨在模拟胃肠道特定部分的生理条件。因此,在这些介质中,pH、渗透压和表面张力与生理值相似(75)。
1986年到1990年,Macheras和Koupparis等人研究了生理相关介质的前体(82,83,84,85,86)。他们用牛奶评估了药物产品的溶出情况。1986年,Macheras、Koupparis和Tsaporonis研究了呋喃妥因、吡罗昔康、吲哚美辛、泼尼松龙、地西泮、双香豆素和灰黄霉素。该介质的使用旨在模拟进食状态的条件。Galia等人发表了第一代生理相关介质FaSSIF和FeSSIF(进食状态模拟肠道)(87)。这些作者于与Machera,Koupparis和Tsaprounis类似,评估了对乙酰氨基酚在牛奶中的溶出情况,证实了先前发表的研究结果。生理相关介质FaSSIF和FeSSIF后来更新,现在被称为FaSSIF-V2和FeSSIF-V2。FaSSIF与FaSSIF-V2之间的主要区别在于FaSSIF-V2中卵磷脂的含量比FaSSIF少(88)。FeSSIF和FeSSIF-V2的区别在于,第二代介质包括两种消化组分:单油酸甘油酯和油酸钠。众所周知,这两种物质都能提高难溶药物的溶解度和溶出度。表II显示了不同代的FaSSIF和FeSSIF的组成(87)。
与水相比,在FaSSIF-V2中,电离和亲脂性对药物的增溶起着重要作用。强碱性药物在FaSSIF-V2中的溶解度增加,可能是由于与负电荷介质具有良好静电相互作用。电离的酸性药物在FaSSIF-V2中的溶解度没有增加(32)。
使用FaSSIF或FaSSIF-V2测定了甲芬那酸、酮康唑和扑热息痛,以及美托洛尔和达那唑等代表性酸性、碱性和中性化合物的溶解度。对于碱性和中性药物,FaSSIF-V2对药物的增溶作用低于FaSSIF。对于在肠道中电离的酸性药物,pH起着更重要的作用。FaSSIF和FaSSIF-V2,这两种空腹状态介质的使用为测定药物溶解度提供了更广泛的空间。市售FaSSIF-V2与文献(88)中描述的用二氯甲烷制备的FaSSIF-V2的结果相同。
研究表明,由于难溶药物的润湿和/或胶束增溶作用增强(26,32,55,89-91),药物生理相关介质中的溶解度比在水中更高(表III)。Yazdanian及其合作者(2004年)研究了非甾体抗炎药(NSAID),结果表明,当用FeSSIF和FaSSIF代替缓冲液0.1 N HCl(pH 1.2)、0.02 M柠檬酸(pH 5.0)和0.02 M Na2HPO4以及0.02 M NaH2PO4(pH 7.4)时,药物溶解度增加(90)。Rinaki等人开发了动态肠道吸收模型,并表明生理相关介质中的溶解度数据可用于处方开发阶段的指导(92)。
Wei和Löbenberg(55)及Okumu等人(89)的研究表明,卵磷脂和胆盐的纯度影响药物溶解度,如格列本脲(补充材料4)和孟鲁司特钠(补充材料5)(55,89)。当卵磷脂和胆盐的纯度较低时,格列本脲的溶解度较高,而当使用95%以上的卵磷脂和牛磺胆酸钠时,孟鲁司特钠的溶解度较高。
最近,Fuchs和Dressman(2014)(93)对生理相关介质和人体肠液(HIF)的组成和理化性质进行了综述。报告指出,必须考虑用于组成生理相关介质的磷脂和胆盐的类型。作者建议应加入游离脂肪酸作为卵磷脂的脂肪分解产物。加入低浓度的胆固醇也可以。
Khoshakhlagh等人(94)发表了此类媒体的一个例子。他们的结论是,在FaSSIF中加入胆固醇会产生生理适应的模型流体FaSSIF-C,增加了难溶性药物的溶解度。FaSSIF-C的成分可根据性别调整。
目前,有许多生理相关介质可代表胃肠道的不同部分。其范围从胃到结肠。
将空腹和进食状态的模拟胃液作为生理相关介质,模拟药物的溶出行为,使药物在进食或不进食时均可在胃内溶解。这些介质的组成见表IV(95)。
表IV. 空腹和进食状态下的模拟胃液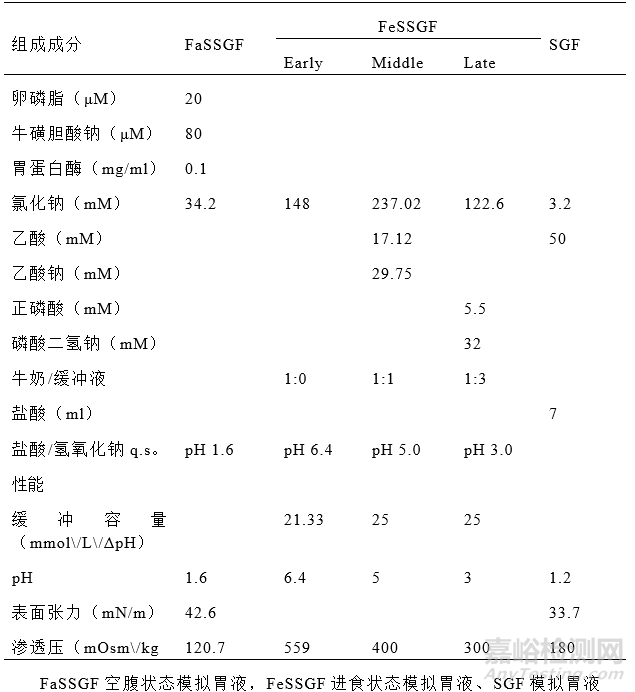
Fotaki等人(96)和Chen等人(97)发表了模拟结肠液(SCoF)的研究(表V)。区别在于缓冲体系。SCoF1采用磷酸盐缓冲液,而SCoF2采用乙酸缓冲液。
表V. 模拟结肠液1(SCoF1)和模拟结肠液2(SCoF2)的组成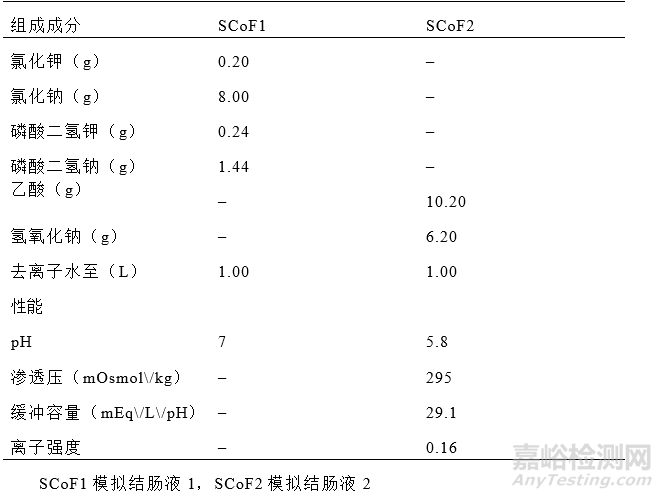
最近一项研究,比较了17种模型药物(补充材料6)在模拟生理相关介质和真实人体肠道液体中的溶解度。该研究分别比较了空腹HIF,进食状态含空白FaSSIF和FeSSIF的人体肠液(HIF),FaSSIF和FeSSIF(33)。用粗(劣质)牛磺胆酸盐制备FaSSIF和FeSSIF。比较表明,模拟介质是用于肠道溶解度评估的生理相关介质,在评估难溶性药物时,与简单缓冲液相比,模拟介质可更好的评估。他们还在磷酸盐缓冲液中测试了不同浓度的聚乙二醇1000维生素E琥珀酸酯(TPGS)。相关性不如FaSSIF和FeSSIF(33)。
目前的文献综述中,56项研究中有12项研究使用了生理相关介质(21.4%)。选择的生理相关介质如下:FaSSIF(21,29,30,33,36,40,44,48,54,57);FeSSIF(21、29、33、40、48、54);空腹和进食HIF(33);LQ FaSSIF(55);以及FaSSIF-V2(32)。
其他研究,与模拟胃液相比,使用含有不同脂肪含量、蛋白质、碳水化合物和氨基酸的牛奶为介质模拟消化的食物,并添加胆汁成分,以提高伊曲康唑溶解度(26)。
正如所见,不同的生理相关介质可用于评估药物在胃肠道不同部分的溶解度。然而,它们的纯度和批次间的一致性存在差异。高纯度的胆盐价格昂贵。此外,由于其复杂的组成,处理耗时较长和步骤复杂,对分析构成了挑战(98)。然而,生理相关介质似乎是评估难溶药物体内溶解度和体内释放行为的最佳起点。
药物溶出度
1897年,Noyes和Whitney发现,固体物质在其自身溶液中的溶解程度与该溶液的浓度和饱和溶液的浓度之差成正比(99)。如今,溶出度测试被用于药品性能测试,以确定药物从某个剂量(如片剂)中释放的速度和程度。因此,介质的选择必须以科学知识和经验为基础(100)。在这篇综述中,我们重点介绍溶出度测试中的介质演变。
水
根据USP第711章(101),当以纯水作为溶出介质而不用作溶剂来测定药物溶解度时,需要对其进行处理以减少溶解的空气。USP第1092章(102)中提到了脱氧水,建议的脱氧方法包括将溶出介质加热到41℃,然后通过0.45μm微孔滤膜真空过滤,并在保持真空的同时用力搅拌滤液。该综述显示,20项研究中的6项(9,14,16,22,23,34)以水为溶出介质。只有1项研究提到了脱气(14)。
水作为溶出介质有一些缺点。例如,由于原料或辅料的影响,其性质在试验过程中会发生变化。此外,水不能为难溶药物提供漏槽条件。漏槽条件定义为获得被测药物剂量的饱和溶液所需体积的至少三倍(103)。制药行业,溶出度测定历来被用作批间药物释放的质量控制方法和评估不同处方的开发工具(104)。
碳酸氢盐缓冲液
最近,由于碳酸氢盐缓冲液与生理缓冲体系的相似性,碳酸氢盐缓冲液的使用引起了人们的兴趣。所用的pH6.8生理性碳酸氢盐缓冲液由Hank’s缓冲液衍生和改性而来,可以区分片剂的肠溶包衣。与药典磷酸盐缓冲液中快速的体外释放相比,碳酸盐缓冲液可区分不同包衣材料之间药物释放的显著差异,这与崩解结果相关(105)。
Varum等人(106)引入了一种碳酸氢盐缓冲液,它是基于Hank’s的缓冲液。pH 5.6的缓冲液很不稳定,必须由Auto pH™系统来维持。该缓冲液是模仿近端小肠的条件。研究表明,该缓冲液可预测不同包衣制剂的体外溶出行为,而磷酸盐缓冲液则不能(补充材料7)。
同样,美沙拉嗪肠溶片的溶出行为表明,与药典磷酸盐缓冲液相比,碳酸氢盐缓冲液可以提高预测能力(107)。Boni等人(108)强调,碳酸氢盐缓冲液必须是新制备的,才能产生可重复的溶出曲线。这可能是由于碳酸氢盐和碳酸的浓度、溶出和溶解的二氧化碳量以及环境中的二氧化碳分压之间复杂而动态的相互作用。这些性质可在生理范围内模拟管腔内pH的动态变化。这通过单个实验完成,不需要通过自动系统改变溶液的离子强度(109)。
如前所述,Krieg等人(77)比较了碳酸盐和磷酸盐缓冲液及其对弱酸和弱碱体外溶出的相关性。得出结论:可能低浓度磷酸盐缓冲液(1-25mM)往往更具生理学意义,更好地模拟碳酸氢盐缓冲液对弱酸性药物溶出的影响。弱碱药物需要极低的磷酸盐缓冲液浓度(<2 mM)来模拟生理相关的碳酸氢盐缓冲液。他们指出,目前使用的磷酸盐缓冲液的盐浓度太高,与体内存在的碳酸盐缓冲强度不相关。作者建议遵循低缓冲浓度,以更好地了解磷酸盐缓冲强度对溶出的影响。
生理相关介质
生理相关介质不仅用于确定药物的溶解度,还用于进行溶出度试验。然而,其性质、价格和成分的可变性使其不能用作常规质量控制介质。研究表明,与其他介质相比,用生理相关介质对难溶性药物进行溶出试验似乎能够更好地模拟体内溶出度(55,85,110–112)。如果体外溶出度与体内溶出度相关,则计算机模拟可能会使用溶出曲线作为药代动力学模型的输入函数。这首先应该考虑建立体外/体内相关性(IVIVC)。Jantraid等人(110)发表了动态溶解条件和生理相关介质与软件应用相结合的体内外相关性的例子。他们用FaSSIF和FeSSIF确定了食物对双氯芬酸钠缓释微丸的影响。此外,若使用流通池,吸收的剂量与溶解剂量之间的相关性更好。
Wei和Löbenberg(55)表明,生理相关介质中pH动态变化会影响格列本脲片的溶出行为,将溶出数据用作模拟软件的输入函数时,得到了类似的结果。溶出度数据可用GasterPlus™软件预测体内的临床数据™, 该软件使用先进的间隔吸收和转运模型。Sunesen等人(111)使用生理相关介质研究了达那唑在空腹和进食条件下的行为,发现使用流通池溶出法,可实现体内外相关性。Okumo等人(89)表明,溶出介质的动态pH变化引起了孟鲁司特钠片的释放曲线的变化,将其用作GastroPlus软件的输入函数时,可预测体内的药代动力学。Fang等人(113)对动态溶出方案进行了微调,并将其应用于食物效应研究,作为早期药物开发的筛选工具。
然而,FaSSIF和FeSSIF并不总是具有预测性。例如,这些介质并未预测出灰黄霉素的吸收和体内行为(54)。为了更好地模拟难溶药物的体内溶出行为,提出了一种新的体外释放介质MREVID2。与FaSSIF相比,该介质中牛磺胆酸钠和磷脂酰胆碱的含量高出7.5倍。灰黄霉素在FaSSIF中的最大药物浓度为9.15μg/ml,是MREVID-2的3.8倍(35.42μg/m L)(54)。
表面活性剂
目前,像很多监管机构,如FDA 2015(114),要求产品规格注重临床相关性,包括溶出规格。但由于生理相关介质的性质,它们在常规质量控制中并不实用,必须考虑其他表面活性剂。表面活性剂的选择和类型基于它们能促进药物溶解,并增加体内的可预测性。
表面活性剂的理化特性、介质的离子强度和缓冲体系的性质取决于研究的药物类型,例如甲芬那酸。当使用十二烷基硫酸钠(SLS)时,该药物的溶解度受离子强度变化的影响。而使用十六烷基三甲基溴化铵(CTAB)则无影响。CTAB、SLS和聚山梨醇酯80是常用的阳离子、阴离子和非离子表面活性剂(表VI)(115)。
表VI. 溶出度测试中常用的表面活性剂
Crison等人(116)描述了用一个简单的相加模型估算吡罗昔康(PX)的溶出度和溶解度的实验,该模型考察了pH和表面活性剂SLS 0.5、1.0和2.0(w/v%)的影响。溶出速率与载药胶束的扩散率成正比,胶束形成的任何变化都会影响溶出过程。药物的总溶解度由各组分的和决定,即Px、Px−和[Px]胶束。该模型能够预测可电离的难溶性药物的溶出度和溶解度随pH和表面活性剂浓度的变化(5)。此外,还必须研究表面活性剂的纯度,因为它对胶束的大小和载药能力有很大影响,这会导致溶解度和溶出速率的变化(117)。
由此可见,表面活性剂的纯度是获得可重复结果的重要质量属性。通常,聚山梨醇酯80每分子含有约20个聚氧乙烯(POE)。然而,其合成过程不仅产生单酯,还产生一些副产物。对聚山梨醇酯80进行了反相高效液相色谱和质谱分析,结果表明聚山梨醇酯80是一种含有POE基团的复杂的聚合物混合物。该表面活性剂不仅含有聚氧乙烯山梨醇单油酸酯(PSM),还含有许多POE中间体,如聚氧乙烯山梨醇(PS)、聚氧乙烯山梨醇二油酸酯(PSD)、聚氧乙烯山梨醇三油酸酯(PStri)和聚氧乙烯山梨醇四油酸酯(PSTetra)(118)。此外,脂肪酸组成为约70%的油酸和其他几种脂肪酸,其纯度也并不总是相同的。因此,应考虑使用纯度更高、成分控制更好的其他表面活性剂进行溶出度测试。
聚氧乙二醇十二烷基醚,商业上称为Brij 35,在一些研究中用作溶出度测试的表面活性剂(95)。该表面活性剂的疏水段与聚山梨醇酯的疏水段具有相似的大小和结构,但Brij 35具有非支化的亲水PEO链,并只有一个单个长链脂肪酸。通过选择(OE)基团的数量与碳氢链的长度,可找出溶出方法开发中表面活性剂性质。避免使用聚山梨醇酯表面活性剂带来的溶出不可重复性(119)。
在预测溶出测试中,表面活性剂组成的胶束模拟了小肠中的胆酸聚集体。表面活性剂促进了溶质向溶剂的扩散和运输。由于溶出是溶解性和扩散性的综合效应,当使用不同的表面活性剂时,胶束大小将影响分子的溶出速率(120)。
生物药剂学系统和溶解度的监管观点
生物药剂学系统(BCS)确定特定药物剂量在胃肠道pH范围内在250ml介质中的溶解度。250ml是指口服给药时同剂型服用的水的体积(121)。
BCS假设,如果某一剂量的药物在生理pH范围内溶于250ml或更少的水中,该药物是高度溶解的。给药后,药物将立即溶解(122,123)。
不同的FDA指导文件使用不同的pH范围来确定药物的溶解度:2000年的生物豁免指南确定了在37±1°C(124)下pH范围为1–7.5时,最高剂量浓度在250 ml或更少的水性介质中的溶解度,而1997年的CEDER指南使用pH1.0–8.0(69),2015年FDA关于含BCS 1类和3类药物的速释固体口服制剂的溶出测试和规范标准的指南草案使用pH范围为1-6.8(113)。
其他全球指导文件,如巴西卫生监督局(ANVISA)和加拿大卫生部指导使用pH范围为1.2-6.8(125,126)。多年来,该pH范围被公认为最合适的pH范围,最能反映胃肠道体内情况。现今不同指导文件使均用250ml介质,但不幸的是,定义测试的剂量不同。
2000年FDA指南(124)和2015年指南草案(127)规定最高(剂量)规格在250 ml水中的溶解度(67,111,119,120)。EMA指南文件(128)、世界卫生组织技术报告(2015)(129)和加拿大卫生部(126)要求也规定了最高单剂量给药的溶解度,特定情况下,最高单剂量可以是250ml中的两个或多个剂量单位(115)。这将许多药物归入“难溶性”药物。
Sediq等人(130)使用新的剂量定义评估了27种药物的剂量-溶解度比(D/S)(以体积ml表示)对BCS分类的影响。国际制药联合会(FIP)在2015年之前已出版了生物豁免专论,因此需要对某些药物进行重新分类。FIP汇编目前包含40多个专论(131)。对其中22种药物,标准的改变并没有改变其BCS分类及生物豁免建议。然而,对于乙酰唑胺、盐酸甲氧氯普胺、盐酸维拉帕米、泼尼松龙和强的松等药物,其最高单次给药剂量如下:500mg(D/S=406ml)、20mg(D/S=472 ml)、240 mg(D/S=500 ml)、100 mg(D/S=412 ml)和100 mg(D/S=752 ml)。溶解剂量所需的体积大于250ml。这些结果改变了这些药物的BCS分类,也改变了其生物豁免的资格。作者的结论是,对于接近溶解度极限(250ml)的药物,需要逐案分析。使用EMA标准可能会将I类药物的分类改为II类,将III类药物的分类改为IV类,使其不符合生物豁免要求(130)。生物等效性研究通常不是比较最高给药剂量,而是比较最大规格。
因此,Daousani和Macheras(132)建议在EMA的《产品特性摘要》(133)中推荐给药的最高单次口服剂量。由于溶出动力学是剂量依赖的,所以溶出度要求(在指定时间内溶解的百分比)可能与FDA和EMA使用的剂量概念不同(132)。作者认为,以实际使用的剂量进行溶出度试验,以指导生物等效性研究的剂量选择标准,在科学上是可以接受的。
2015年修改后的WHO标准(134)认为,最高单次治疗剂量在37±1°C下,在pH为1.2–6.8的250 ml或更少的水介质中可溶时,即为高溶性药物。根据这份新报告,符合生物豁免的药物必须以最高的上市规格进行体内生物等效性研究。
另一方面,Yazdanian等人(90)认为,FDA对弱酸的溶解度标准过于严格,因为酸在胃中溶解度不高,但在肠道中溶解度很高,而肠道是药物吸收的场所。根据其研究,采用pH 1.2-7.4的溶解度标准,18种酸性非甾体抗炎药中的15种会被归入2类化合物。仅采用pH7.4的溶解度标准,有15种药物被归为1类药物。作者建议,根据BCS(90)对乙酸化合物进行分类时,pH溶解度范围应在5.0–7.4之间。这项研究的基本原理已纳入2006年WHO的生物豁免指南(129),该指南允许对弱酸进行生物豁免。遗憾的是,这一点在2015年的指南中被删除(134)。
溶解度标准是BCS对药物进行分类的两个参数之一。目前,根据WHO的基本药物清单,阿莫西林是BCS I类药物(135,136)。由于最高剂量标准,EMA将其列为BCS 2类药物,而由于渗透性标准的不同,FDA将其列为BCS 4类药物。
总的来说,BCS是一种口服生物利用度的科学方法,并作为一种科学风险管理工具引入监管科学,在某些条件下豁免生物等效性研究,如FDA SUPAC指南(137)首次所做的那样。不幸的是,从那时起,不同的指南引入了不同的定义。如阿莫西林那样,这给全球运营公司带来更多的困惑。
正如所述,不同的指导文件之间有统一的空间,以便为全球药物制定相同的BCS类别,因为溶解度不会因为在不同国家确定而改变。
最后的考虑
本文介绍了测定药物溶出度的介质和缓冲液的发展,以及预测溶出介质的发展趋势。作为生理相关介质的潜在替代品的表面活性剂的类型必须仔细考虑。此时,没有通用的介质可以用来检测药物的溶解度或药物在体内的溶出行为。然而,为了优化溶出介质的体内可预测性,已对其进行了许多改进。正确选择溶出介质来预测临床相关剂型属性仍然是在个案的基础上进行。此外,溶出装置也会对溶出度曲线的形状产生影响;然而,这不是本次综述的部分。虽然几年前,为每个药物分子建立体内外相关性似乎是不可能的,但目前在介质组成、溶出装置设计和计算机模拟方面的所有发展都表明,这在未来是可能的。过去已经发布了此类综合软件指导方法的例子(55,85,110,138)。然而,对体外和体内溶出过程的精确机制理解对于制定临床相关溶出规范并将体外溶出与临床结果联系起来至关重要。
参考文献
1. Aulton M, Taylor K. Aulton’s pharmaceutics: The design and manufacture of medicines. 4th ed. London: Churchill Livingstone; 2013.
2. Higuchi T, Connors K. Phase-solubility techniques. Adv Anal Chem Instrum. 1965; 7:117-212.
3. Jouyban A. Handbook of solubility data for pharmaceuticals. Boca Raton: CRC Press; 2009.
4. United States Pharmacopeial Convention. USP 38-NF 33, General notices and requirements. Rockville: United States Pharmacopeial Convention; 2015.
5. Jinno J, Oh D, Crison JR, Amidon GL. Dissolution of ionizable water-insoluble drugs: the combined effect of pH and surfactant. J Pharm Sci. 2000;89(2):268-74.
6. Arya P, Pathak K. Assessing the viability of microsponges as gastro retentive drug delivery system of curcumin: optimization and pharmacokinetics. Int J Pharm 2014;460(1-2):1-12.
7. Mota FL, Carneiro AP, Queimada AJ, Pinho SP, Macedo EA. Temperature and solvent effects in the solubility of some pharmaceutical compounds: measurements and modeling. Eur J Pharm Sci. 2009;37(3-4):499-507.
8. Maulvi FA, Dalwadi SJ, Thakkar VT, Soni TG, Gohel MC, Gandhi TR. Improvement of dissolution rate of aceclofenac by solid dispersion technique. Powder Technol. 2011;207(1-3):47-54.
9. Goud NR, Suresh K, Nangia A. Solubility and stability advantage of aceclofenac salts. Cryst Growth Des.2013;13(4):1590-601.
10. Kommavarapu P, Maruthapillai A, Palanisamy K, Sunkara M. Preparation and characterization of rilpivirine solid dispersions with the application of enhanced solubility and dissolution rate. Beni-Suef Univ J Basic Appl Sci. 2015;4(1):71-9.
11. Persson AM, Pettersson C, Sokolowski A. Correlation of in vitro dissolution rate and apparent solubility in buffered media using a miniaturized rotating disk equipment: part II. Comparing different buffer media. Drug Discov Ther. 2009;3(3):114-22.
12. Florindo C, Araújo JMM, Alves F, Matos C, Ferraz R, Branco L, et al. Evaluation of solubility and partition properties of ampicillin-based ionic liquids. Int J Pharm. 2013; 456(2):553-9.
13. Tomaszewska I, Karki S, Shur J, Price R, Fotaki N. Pharmaceutical characterisation and evaluation of cocrystals: importance of in vitro dissolution conditions and type of coformer. Int J Pharm. 2013;453(2):380-8.
14. Grossjohann C, Eccles KS, Maguire AR, Lawrence SE, Tajber L, Corrigan OI, et al. Characterisation, Solubility and intrinsic dissolution behaviour of benzamide: dibenzyl sulfoxide cocrystal. Int J Pharm. 2012;422(1-2):24-32.
15. Prabakaran D. Effect of hydrophilic polymers on the release of diltiazem hydrochloride from elementary osmotic pumps. Int J Pharm. 2003;259(1-2):173-9.
16. Sehić S, Betz G, Hadzidedić S, El-Arini SK, Leuenberger H. Investigation of intrinsic dissolution behavior of different carbamazepine samples. Int J Pharm. 2010;386(1-2):77-90.
17. Barzegar-Jalali M, V alizadeh H, Shadbad M-RS, Adibkia K, Mohammadi G, Farahani A, et al. Cogrinding as an approach to enhance dissolution rate of a poorly water-soluble drug (gliclazide). Powder Technol. 2010;197(3):150-8.
18. Srirangam R, Majumdar S. Passive asymmetric transport of hesperetin across isolated rabbit cornea. Int J Pharm. 2010;394(1-2):60-7.
19. Völgyi G, Baka E, Box KJ, Comer JEA, Takács-Novák K. Study of pH-dependent solubility of organic bases. Revisit of Henderson-Hasselbalch relationship. Anal Chim Acta.2010;673(1):40-6.
20. Gao Y, Gao J, Liu Z, Kan H, Zu H, Sun W, et al. Coformer selection based on degradation pathway of drugs: a case study of adefovir dipivoxil-saccharin and adefovir dipivoxil nicotinamide cocrystals. Int J Pharm. 2012;438(1-2):327-35.
21. Takács-Novák K, Szőke V, Völgyi G, Horváth P, Ambrus R, Szabó-Révész P. Biorelevant solubility of poorly soluble drugs: rivaroxaban, furosemide, papaverine and niflumic acid. J Pharm Biomed Anal. 2013; 83:279-85.
22. Agrawal S, Panchagnula R. Dissolution test as a surrogate for quality evaluation of rifampicin containing fixed dose combination formulations. Int J Pharm. 2004;287(1-2):97-112.
23. Li S, Wong S, Sethia S, Almoazen H, Joshi YM, Serajuddin A TM. Investigation of solubility and dissolution of a free base and two different salt forms as a function of pH. Pharm Res.2005;22(4):628-35.
24. Bartolomei M, Bertocchi P, Antoniella E, Rodomonte A. Physico-chemical characterisation and intrinsic dissolution studies of a new hydrate form of diclofenac sodium: comparison with anhydrous form. J Pharm Biomed Anal. 2006;40(5):1105-13.
25. Baka E, Comer JEA, Takács-Novák K. Study of equilibrium solubility measurement by saturation shake-flask method using hydrochlorothiazide as model compound. J Pharm Biomed Anal. 2008;46(2):335-41.
26. Ghazal HS, Dyas AM, Ford JL, Hutcheon GA. In vitro evaluation of the dissolution behaviour of itraconazole in biorelevant media. Int J Pharm. 2009;366(1-2):117-23.
27. Sheng JJ, Kasim NA, Chandrasekharan R, Amidon GL. Solubilization and dissolution of insoluble weak acid, ketoprofen: effects of pH combined with surfactant. Eur J Pharm Sci. 2006;29(3-4):306-14.
28. Loftsson T, Matth K, Másson M. The effects of organic salts on the cyclodextrin solubilization of drugs. Int J Pharm.2003; 262:101-7.
29. Bard B, Martel S, Carrupt P. High throughput UV method for the estimation of thermodynamic solubility and the determination of the solubility in biorelevant media. Eur J Pharm Sci. 2007; 3:230-40.
30. Vogt M, Kunath K, Dressman JB. Dissolution improvement of four poorly water soluble drugs by cogrinding with commonly used excipients. Eur J Pharm Biopharm. 2008;68(2):330-7.
31.Charkoftaki G, Kytariolos J, Macheras P. Novel milk-based oral formulations: proof of concept. Int J Pharm. 2010;390(2):150-9.
32. Ottaviani G, Gosling DJ, Patissier C, Rodde S, Zhou L, Faller B. What is modulating solubility in simulated intestinal fluids? Eur J Pharm Sci. 2010;41(3-4):452-7.
33. Clarysse S, Brouwers J, Tack J, Annaert P, Augustijns P. Intestinal drug solubility estimation based on simulated intestinal fluids: comparison with solubility in human intestinal fluids. Eur J Pharm Sci. 2011;43(4):260-9.
34. Kolašinac N, Kachrimanis K, Homšek I, Grujić B, Ðurić Z, Ibrić S. Solubility enhancement of desloratadine by solid dispersion in poloxamers. Int J Pharm. 2012;436(1-2):161-70.
35. Guhmann M, Preis M, Gerber F, Pöllinger N, Breitkreutz J, Weitschies W. Development of oral taste masked diclofenac formulations using a taste sensing system. Int J Pharm. 2012;438(1-2):81-90.
36. Niederquell A, Kuentz M. Biorelevant dissolution of poorly soluble weak acids studied by UV imaging reveals ranges of fractal-like kinetics. Int J Pharm. 2014;463(1):38-49.
37. Thing M, Jensen SS, Larsen C, Østergaard J, Larsen SW. Modification of concomitant drug release from oil vehicles using drug-prodrug combinations to achieve sustained balanced analgesia after joint installation. Int J Pharm. 2012;439(1-2):246-53.
38. Baek J, Lim J, Kang J, Shin S, Jung S, Cho C. Enhanced transdermal drug delivery of zaltoprofen using a novel formulation. Int J Pharm. 2013;453(2):358-62.
39. Box KJ, Völgyi G, Baka E, Stuart M, Takács-Novák K, Comer JE. Equilibrium versus kinetic measurements of aqueous solubility, and the ability of compounds to supersaturate in solution—a validation study. J Pharm Sci. 2006; 95:1298-307.
40. Do TT, V an Speybroeck M, Mols R, Annaert P, Martens J, V an Humbeeck J, et al. Theconflict between in vitro release studies in human biorelevant media and the in vivo exposure in rats of the lipophilic compound fenofibrate. Int J Pharm. 2011;414(1-2):118-24.
41. Dokoumetzidis A, Papadopoulou V, V alsami G, Macheras P. Development of a reaction-limited model of dissolution: application to official dissolution tests experiments. Int J Pharm. 2008; 355:114-25.
42. Dressman JB, Nair A, Abrahamsson B, Barends DM, Groot DW, Kopp S, et al. Biowaiver monograph for immediaterelease solid oral dosage forms: acetylsalicylic acid. J Pharm Sci. 2012; 101:2653-67.
43. El-Gendy AM, Adejare A. Membrane permeability related physicochemical properties of a novel gamma-secretase inhibitor. Int J Pharm. 2004;280(1-2):47-55.
44. Fischer SM, Parmentier J, Buckley ST, Reimold I, Brandl M, Fricker G. Oral bioavailability of ketoprofen in suspension and solution formulations in rats: the influence of poloxamer 188. J Pharm Pharmacol. 2012;64(11):1631-7.
45. Gao Y, Liao J, Qi X, Zhang J. Coamorphous repaglinide saccharin with enhanced dissolution. Int J Pharm. 2013;450(1-2):290-5.
46. Kaur M, Kaur R, Pissurlenkar RRS, Coutinho EC, Kumar U,Prakash O, et al. Telmisartan complex augments solubility, dissolution and drug delivery in prostate cancer cells. Carbohydr Polym. 2014; 101:614-22.
47. Kumar L, Jog R, Singh S, Bansal A. Effect of counterion on the solid state photodegradation behavior of prazosin salts. AAPS PharmSciTech. 2013;14(2):757-63.
48. Levis KA, Lane ME, Corrigan OI. Effect of buffer media composition on the solubility and effective permeability coefficient of ibuprofen. Int J Pharm. 2003;253(1-2):49-59.
49. Nair A, Abrahamsson B, Barends DM, Groot DW, Kopp S, Polli JE, et al. Biowaiver monographs for immediate release solid oral dosage forms: amodiaquine hydrochloride. 2012;101(12):4390-401.
50. Nokhodchi A, Shokri J, Dashbolaghi A, Hassan-Zadeh D, Ghafourian T, Barzegar-Jalali M. The enhancement effect of surfactants on the penetration of lorazepam through rat skin. Int J Pharm. 2003;250(2):359-69.
51.Zakeri-Milani P, Barzegar-Jalali M, Azimi M, V alizadeh H. Biopharmaceutical classification of drugs using intrinsic dissolution rate (IDR) and rat intestinal permeability. Eur J Pharm Biopharm. 2009;73(1):102-6.
52. Avdeef A, Tsinman O. Miniaturized rotating disk intrinsic dissolution rate measurement: effects of buffer capacity in comparisons to traditional wood’s apparatus. Pharm Res. 2008;25(11):2613-27.
53. Berger CM, Tsinman O, V oloboy D, Lipp D, Stones S, Avdeef A. Technical note: miniaturized intrinsic of griseofulvin and carbamazepine. Dissolution Technol. 2007;(1):39-41.
54. Fujioka Y, Kadono K, Fujie Y, Metsugi Y, Ogawara K, Higaki K, et al. Prediction of oral absorption of griseofulvin, a BCS class II drug, based on GITA model: utilization of a more suitable medium for in-vitro dissolution study. J ControlRelease. 2007;119(2):222-8.
55. Wei H, Löbenberg R. Biorelevant dissolution media as a predictive tool for glyburide a class II drug. Eur J Pharm Sci.2006;29(1):45-52.
56. Y u LX, Carlin AS, Amidon GL, Hussain AS. Feasibility studies of utilizing disk intrinsic dissolution rate to classify drugs. Int J Pharm. 2004; 270:221-7.
57. Fong SYK, Ibisogly A, Bauer-Brandl A. Solubility enhancement of BCS class II drug by solid phospholipid dispersions: spray drying versus freeze-drying. Int J Pharm. 2015;496(2):382-91.
58. Marano S, Barker SA, Raimi-Abraham BT, Missaghi S, Rajabi-Siahboomi A, Craig DQM. Development of microfibrous solid dispersions of poorly water-soluble drugs in sucrose using temperature-controlled centrifugal spinning. Eur J Pharm Biopharm. 2016; 103:84-94.
59. Kambayashi A, Yasuji T, Dressman JB. Prediction of the precipitation profiles of weak base drugs in the small intestine using a simplified transfer (Bdumping^) model coupled with in silico modeling and simulation approach. Eur J Pharm Biopharm. 2016; 103:95-103.
60. Bergström CAS, Luthman K, Artursson P. Accuracy of calculated pH-dependent aqueous drug solubility. Eur J Pharm Sci. 2004;22(5):387-98.
61. Völgyi G, Marosi A, Takács-Novák K, Avdeef A. Salt solubility products of diprenorphine hydrochloride, codeine and lidocaine hydrochlorides and phosphates—novel method of data analysis not dependent on explicit solubility equations. ADMET DMPK. 2013;1(4):48-62.
62. Avdeef A. Anomalous solubility behavior of several acidic drugs. ADMET DMPK. 2014;2(1):33-42.
63. Fini A. Solubility and solubilization properties of non-steroidal anti-inflammatory drugs. Int J Pharm. 1995;126(1-2):95-102.
64. Higuchi T, Gupta M, Busse LW. Influence of electrolytes, pH, and alcohol concentration on the solubilities of acidic drugs. J Am Pharm Assoc (Scientific ed). Wiley Subscription Services, Inc., A Wiley Company; 1953 Mar;42(3):157-61.
65. Fadda HM, Merchant HA, Arafat BT, Basit AW. Physiological bicarbonate buffers: stabilisation and use as dissolution media for modified release systems. Int J Pharm. 2009;382(1-2):56-60.
66. Klein S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010;12(3):397-406.
67. Li Z, He X. Physiologically based in vitro models to predict the oral dissolution and absorption of a solid drug delivery system. Curr Drug Metab. 2015;16(9):777-806.
68. U.S. Food and Drug Administration. Guidance for Industry: Dissolution testing of immediate release solid oral dosage forms. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research(CDER),1997.http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ ucm070237.pdf. Accessed 11 Nov 2016.
69. Dressman JB, V ertzoni M, Goumas K, Reppas C. Estimating drug solubility in the gastrointestinal tract. Adv Drug Deliv Rev. 2007;59(7):591-602.
70. United States Pharmacopeial Convention. USP 38-NF 33, drug release <724>. Rockville: United States Pharmacopeial Convention; 2015
71. United States Pharmacopeial Convention. USP 40-NF 35, Update: Dissolution methods database. Rockville: United States Pharmacopeial Convention; 2017. http://www.usp.org/sites/default/files/usp_pdf/EN/USPNF/compendialTools/ dissolution_methods_database.xlsx. Accessed 20 Nov 2016.
72. Stippler E, Kopp S, Dressman JB. Comparison of US Pharmacopeia simulated intestinal fluid TS (without pancreatin) and phosphate standard buffer pH 6.8, TS of the International Pharmacopoeia with respect to their use in in vitro dissolution testing. Dissolution Technol. 2004;11(2):6-10.
73. Almukainzi M, Salehi M, Bou-Chacra N, Löbenberg R. Investigation of the performance of the disintegration test for dietary supplements. AAPS J. 2010;12(4):602-7.
74. Ropers MH, Czichocki G, Brezesinski G. Counterion effect on the thermodynamics of micellization of alkyl sulfates. J Phys Chem B Am Chem Soc. 2003;107(22):5281-8.
75. Khan MZI, Rausl D, Radosević S, Filić D, Danilovski A, Dumić M, et al. Classification of torasemide based on the Biopharmaceutics Classification System and evaluation of the FDA biowaiver provision for generic products of class I drugs. J Pharm Pharmacol. 2006;58(11):1475-82.
76.Jantratid E, Janssen N, Reppas C, Dressman JB. Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update. Pharm Res. 2008;25(7):1663-76.
77.Krieg BJ, Taghavi SM, Amidon GL, Amidon GE. In vivo predictive dissolution: comparing the effect of bicarbonate and phosphate buffer on the dissolution of weak acids and weak bases. J Pharm Sci. 2015;104(9):2894-904.
78. Rangel-Y agui CO, Pessoa A, Tavares LC. Micellar solubilization of drugs. J Pharm Pharm Sci. 2005;8(2):147-63.
79. Zangenberg NH, Müllertz A, Gjelstrup Kristensen H, Hovgaard L. A dynamic in vitro lipolysis model. Eur J Pharm Sci. 2001;14(3):237-44.
80. Ottaviani G, Wendelspiess S, Alvarez-Sánchez R. Importance of critical micellar concentration for the prediction of solubility enhancement in biorelevant media. Mol Pharm. 2015;12(4):1171-9.
81. Kaukonen AM, Boyd BJ, Porter CJH, Charman WN. Drug solubilization behavior during in vitro digestion of simple triglyceride lipid solution formulations. Pharm Res. 2004;21(2):245-53.
88. Monitoring and Control of Genotoxic Impurity Acetamide in the Synthesis of Zaurategrast Sulfate. Org. Process Res. Dev. 2010, 14 (4), 1008−1014.
89. Re-evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 71; International Agency for Research on Cancer, 1999.
90. European Chemicals Agency. Registration Dossier for Acetamide.https://echa.europa.eu/registration-dossier/-/registered-dossier/17464/7/7/1 (accessed 2020-07-15).
91. The Food Contaminant Acetamide Is Not an in VivoClastogen, Aneugen, or Mutagen in Rodent Hematopoietic Tissue. Regul. Toxicol. Pharmacol. 2019, 108, 104451.
92. An Orthogonal Approach for Determination of Acetamide Content in Pharmaceutical Drug Substance and Base-Contaminated Acetonitrile by GC and GC-MS External Method. J. Chromatogr. Sci. 2019, 57 (9), 769−777.
93. A selective and sensitive method development and validation by LC−MS/MS approach for trace level quantification of three potential genotoxic impurities in pantoprazole sodium drug substance. Rasayan J. Chem. 2017, 10, 1080−1087.
94. Initial Assessment Report for Acetanilide. OECD SIDS, 2001.https://hpvchemicals.oecd.org/ui/handler.axd?id=5dd3359f-e67e-49ce-92b3-de0706cba0a5 (accessed 2020-07-15).
95. Bergman, K.; Müller, L.; Teigen, S. W. The Genotoxicity and Carcinogenicity of Paracetamol: A Regulatory (Re) View. Mutat. Res., Fundam. Mol. Mech. Mutagen. 1996, 349 (2), 263−288.
96. National Toxicology Program, U.S. DHHS. Genetic Toxicity Evaluation of N-Acetyl-m-aminophenol in Salmonella/E.coli Mutagenicity Test or Ames Test. Study 667110. https://manticore.niehs.nih.gov/cebssearch/study/002-02528-0001-0000-0 (accessed 2020-07-15).
97. Bomhard, E. M.; Herbold, B. A. Genotoxic Activities of Aniline and Its Metabolites and Their Relationship to the Carcinogenicity of Aniline in the Spleen of Rats. Crit. Rev. Toxicol. 2005, 35 (10), 783−835.
98. European Chemicals Agency. Registration Dossier for Propionamide. https://echa.europa.eu/es/registration-dossier/-/registereddossier/30038/7/7/1 (accessed 2020-07-15).
99. Reregistration Eligibility Decision Document TXR 0050210:Propanil. U.S. Environmental Protection Agency, 2001. https://www3.epa.gov/pesticides/chem_search/cleared_reviews/csr_PC-028201_9-Nov-01_a.pdf (accessed 2020-07-15).
100. Snodin, D.; Teasdale, A. Mutagenic Alkyl-Sulfonate Impurities in Sulfonic Acid Salts: Reviewing the Evidence and Challenging Regulatory Perceptions. Org. Process Res. Dev. 2015, 19 (11), 1465−1485.
101. Elusive Impurities-Evidence versus Hypothesis. Technical and Regulatory Update on Alkyl Sulfonates in Sulfonic Acid Salts. Org. Process Res. Dev. 2019, 23 (5), 695−710.

来源:溶出之家


