您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-06-04 17:28
基因毒性杂质(genotoxic impurity,GTI)是指药物中能直接或间接导致DNA受损引起基因突变,并具有致癌性或者潜在致癌可能性的一类杂质。由于其较一般杂质具有微量水平就存在潜在致突变性和致癌性风险的特点,受到药品监管机构和制药企业重点关注和严格控制。
2002年欧洲药物管理局(EMA)最先出台了关于基因毒性杂质的管理法规,美国食品药物管理局(U.S. FDA)、国际人用药品注册技术要求国际协调会(ICH)等组织也针对基因毒性杂质先后颁发了相关界定、分类、限度、检测和风险评估程序等一系列指南,目前已经成为药品获批及上市的关键指标之一。
我国对基因毒杂质的管理要求
国家食品药品监督管理局于2016年发布的《化学药品新注册分类申报资料要求(试行)》(2016年 第80号)中提到过“结合起始原料和本品的制备工艺,简述对原料药可能存在的基因毒性杂质所进行的分析和研究的结果,并按照ICH M7指导原则的要求说明控制的策略”。
我国药监部门于2017年6月加入ICH,后续2020版中国药典四部通则部分,添加了《9306遗传毒性杂质控制指导原则》,该指导原则的内容要求与ICH M7基本一致。
2024年1月5日,国家药品监督管理局发布了决定使用《M7(R2):评估和控制药物中DNA反应性(致突变)杂质以限制潜在致癌风险》国际人用药品注册技术协调会指导原则的公告。国内医药企业对于相关药品遗传毒性杂质的控制也应符合M7(R2)的指导要求。
基因毒性警示结构
所谓警示结构单元是指一些具有与遗传物质发生化学反应能力的特殊结构单元,会诱导基因突变或者导致染色体重排或断裂,从而具有潜在的致癌风险。警示结构单元是遗传毒性杂质识别的起点。具有警示结构单元,但并未经实验测试模型验证的杂质叫做潜在遗传毒性杂质。常见警示结构单元如图1:
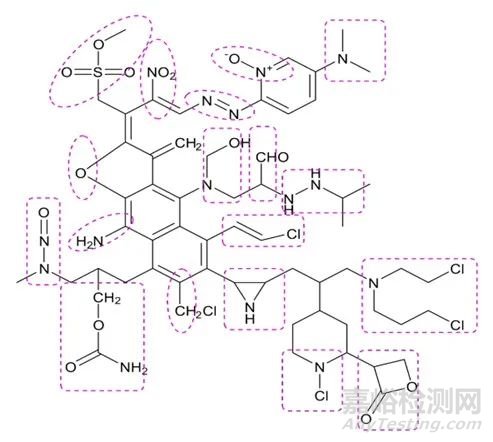
图1 常见警示结构单元
可以发现,以上结构单元大部分都含有氨氮结构,其中亚硝胺类就是比较常见的一种基因毒杂质。
自2018年7月在缬沙坦原料药中检出N-亚硝基二甲胺(NDMA)以来,陆续在其他沙坦类原料药中检出了各类亚硝胺杂质,如NDMA、N-亚硝基二乙胺(NDEA)等。进一步的调查发现,在个别供应商的非沙坦类的药物中(如雷尼替丁),亦有亚硝胺类杂质的检出。亚硝胺类杂质属于ICH M7(R1)(《评估和控制药物中DNA反应性(致突变)杂质以限制潜在致癌风险》)指南[1]中提及的“关注队列”物质。根据世界卫生组织公布的致癌物清单[2],NDMA和NDEA均属于2A类致癌物质;根据国际认可数据库,已有部分亚硝胺类杂质有公开的致癌性数据,如NDMA、NDEA、N-亚硝基-N-甲基-4-氨基丁酸(NMBA)、N-亚硝基二丁胺(NDBA)等。
随着对亚硝胺类杂质的日益关注,美国药典(USP)也在不断更新亚硝胺药物分析杂质,目前总计达到49种,简单亚硝胺和NDSRIs的杂质清单还在持续丰富中。为了保证药品的安全和质量可控,实现有效的风险控制,提供更加可靠的数据支持[3]。
亚硝胺类杂质来源
根据目前所知,亚硝胺类杂质有多种产生原因[4],如工艺产生、降解途径和污染引入等。具体来讲,亚硝胺类杂质可能通过以下途径引入[5]:
(一)由工艺引入亚硝胺类杂质的风险
以沙坦类药物的合成为例,四唑的环化是由有机腈与叠氮化钠的环加成反应在高沸点溶剂(如二甲基甲酰胺或者N-甲基吡咯烷酮)中实现的,反应结束后,过量的叠氮化钠用亚硝酸钠淬灭。然而在这个过程中使用的二甲基甲酰胺和N-甲基吡咯烷酮中分别含有二甲胺和4-甲氨基丁酸杂质(这些杂质即可能是这些溶剂的合成原料,也可能是溶剂分子降解的产物)。这些仲胺在叠氮化钠的淬灭反应中与淬灭剂亚硝酸钠作用,产生了相应的亚硝胺NDMA (二甲基亚硝胺) 和NMBA (N-亚硝基-N-甲基-4-氨基丁酸)。
这个过程是沙坦类药物产生亚硝胺杂质的根本原因(图2)。尤其是考虑到叠氮化钠淬灭是整个沙坦合成工艺的最后一步,产生的亚硝胺杂质污染最终产品的可能性大大增加。
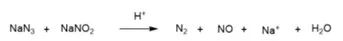
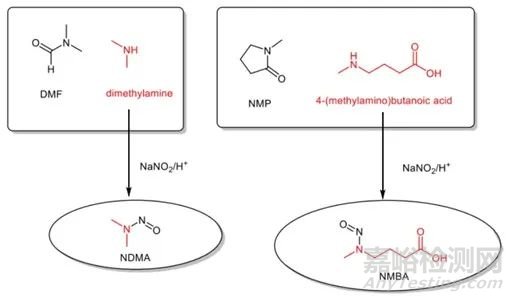
图2 沙坦类药物可能引入亚硝胺类杂质过程示例
(二)由污染引入的风险
原料药生产过程中使用了被亚硝胺类杂质污染的物料(起始物料、中间体、溶剂、试剂、催化剂等)可能带来亚硝胺类杂质的风险。
使用回收的物料亦有引入亚硝胺类杂质的风险。已发现的回收物料被亚硝胺污染的实例包括邻二甲苯、氯化三丁基锡(用作叠氮化三丁基锡的来源)、N,N-二甲基甲酰胺(DMF)。
(三)降解产生风险
某些药物本身会降解产生亚硝胺类杂质,如雷尼替丁在高温下会产生亚硝胺类杂质[6];氨基比林中二甲氨基自身水解也会生成二甲基亚硝胺(图3)。
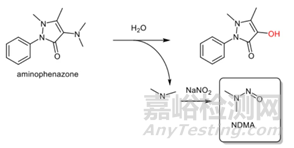
图3 氨基比林降解导致二甲基亚硝胺形成的机理
(四)制剂过程中形成
2019年9月,一份新的二甲基亚硝胺和二乙基亚硝胺产生机理报告被递交到了监管部门。该报告提出,NDMA/NDEA似乎是在盖箔印刷过程中形成的,N-亚硝胺的形成是由盖箔中的硝化纤维素与含胺印刷油墨(二甲胺和二乙胺)反应引起的,并通过汽化和冷凝热封起泡过程转移到在成品上。由于增塑硝化纤维碎片的爆燃温度,在热诱导分解时会产生不同的氮氧化物。从硝酸纤维素中释放出氮氧化物以及随后在印刷药物油墨中对胺进行亚硝化被认为是合理的。
针对以上产生途径,FDA在2024年4月11日-12日组织的年度仿制药论坛中,也给API生产商提出了如下建议:
从整体上评估原料药合成路线,规避亚硝胺类在反应中产生,或将可能性降到最低;
对原材料和中间供应商进行评估和审计,如果发现亚硝胺杂质的风险,应使用高灵敏度且经过验证的方法进行确认;
如果检测到亚硝胺类物质,应进行根本原因分析,必要时变更生产工艺,以防止/减少亚硝胺类物质的生成;
如实向FDA报告进行变更的内容
亚硝胺类基因毒杂质控制策略
(一)基本控制理念
由于亚硝胺类杂质在人体中可接受限度较小,微量杂质的检测和控制难度大。因此对于亚硝胺类杂质的控制应采取避免为主,控制为辅的策略。
1)避免为主是指在药品的研发阶段应根据亚硝胺类杂质产生的原因从原料药工艺路线的选择、物料的选择与质控、工艺条件的优化等方面尽量避免亚硝胺类杂质的产生,并在生产过程中严格执行各操作规范。药品上市许可持有人/药品生产企业应与各物料(原料药应包括起始物料、溶剂、试剂、催化剂、中间体等,制剂应包括原料药、辅料、包材等等)生产商充分沟通,对物料生产和回收工艺进行系统评估。
若评估发现有生成亚硝胺类杂质的风险,应首先分析亚硝酸盐或者可能形成亚硝胺类杂质的相关试剂和溶剂在工艺中使用的必要性,尽量避免选择可能生成亚硝胺类杂质的生产工艺[6]。
2)控制为辅的策略是指当评估药品具有亚硝胺类杂质残留风险且相关工艺无法避免时,应尽可能将该步骤调整至工艺的早期,利用后续多步骤的操作降低亚硝胺类杂质残留风险。同时须根据工艺路线分析可能生成的亚硝胺结构,并优化工艺,制定详细的过程控制策略,保证生产过程中此类杂质的有效去除。
由降解产生亚硝胺类杂质的情况,应分析降解产生的条件,通过优化生产工艺、处方、贮存条件等,降低降解杂质的产生风险。
(二)限度控制
药物中亚硝胺类杂质的控制策略建议参考ICH M7(R1)指南的相关规定,应保证最终拟定的控制策略和杂质限度具有充分合理的科学依据。亚硝胺类杂质的致癌风险较高,不适合按照ICH M7(R1)提出的1.5μg/天的毒理学关注阈值(TTC)控制限度。
1)应使用来自研究设计完善的致癌性试验中的最低TD50值,或与人类风险评估最相关的种属、性别和肿瘤发生器官部位的最低TD50值来计算可接受摄入量,设定对应肿瘤发生风险为十万分之一,人体体重统一按50kg计算,则该亚硝胺类杂质的每日可接受摄入量(Acceptable Intake,AI)为:TD50(mg/kg/天)×50kg/50000。
2)未能在权威机构数据库中查询到TD50值时,可选用以下几种方法分别获得该亚硝胺类杂质的控制限度,并建议取其中最小值:
A.可以参考国际权威机构,如WHO、国际化学品安全性方案(International Programme on Chemical Safety,IPCS)等公布的数据或建立的风险评估方法。
B.与已有TD50值的亚硝胺类杂质结构相似,可以导用其TD50值计算杂质限度。
(三)检测方法的建立
药物中亚硝胺类杂质的分析测试方法,可以参考权威机构发布的方法,亦可自行开发方法,均需注意分析方法灵敏度应与所论证的杂质限度相匹配,并采用杂质对照品进行完整的方法学验证,保证亚硝胺类杂质能够准确有效的检出。若采用自行开发方法,需证明该方法等效于或者更优于同品种官方公布的方法。
(四)全生命周期的风险控制
对于申报上市的产品,申请人在研发中,应进行亚硝胺类杂质的风险评估,对明确有亚硝胺类杂质潜在风险的品种应进行充分的研究,在申报资料的相应章节提交亚硝胺类杂质的研究资料及检测结果,同时应注意用于研究的样品的批次、批量必须具有代表性以及科学依据。
对于已上市药品,药品上市许可持有人/药品生产企业也应主动对于亚硝胺类杂质存在的风险进行评估,若存在潜在的亚硝胺类杂质产生风险,可参照本指导原则以及其他相关指导原则的要求进行研究,根据研究结果采取相应的措施,以防止或最小化患者亚硝胺类杂质的暴露。
参考文献
1. ICH M7(R1) Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals of limit potential carcinogenic risk [EB/OL] (2017-03-31)
2. 世界卫生组织国际癌症研究机构致癌物清单,http://samr.cfda.gov.cn/WS01/CL1991/215896.html
3. 美国药典官方网站, https://www.usp.org/
4. (a) Assessment report, 14 February 2019, EMA/217823/2019. (b)A Screening Procedure for the Formation of Nitroso Derivatives and Mutagens by Drug-Nitrite Interaction, Chem. Pharm. Bull. 1982, 30(9), 3399-3404. (c) Formation of N-Nitrosodimethylamine (NDMA) from Dimethylamine during Chlorination, Environ. Sci. Technol. 2002, 36, 588-595. (d) N-nitrosomethylanlaniline, Org. Synth. 1933, 13, 82. (e) Nitrosomethylurea. Org. Synth. 1935, 15, 48.
5. (a) Information on nitrosamines for marketing authorisation holders,EMA/189634/2019. (b) Questions and answers on “Information on nitrosamines for marketing authorisation holders”,EMA/CHMP/428592/2019 Rev.1
6. Questions and Answers: NDMA impurities in ranitidine (commonly known as Zantac),
https://www.fda.gov/drugs/drug-safety-and-availability/
7. Inhibition of Nitrosamine Formation by Inorganic and Organic Salts, Chem Pharm Bull, 1986, 34(8), 3485-3487.

来源:Internet


