您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-06-08 20:56
摘 要 / Abstract
近年来先进治疗药品研发申报呈爆发式增长,引领生物医药的第三次产业革命。全球范围内嵌合抗原受体T 细胞(CAR-T)治疗产品、腺相关病毒载体基因治疗产品等不同类型的细胞和基因治疗产品相继批准上市,为复发难治疾病提供了有效的治疗手段。先进治疗药品通常采用细胞或组织经基因修饰和(或)体外操作制备,种类包括基因修饰细胞、核酸、病毒载体和人工组织等。由于这类产品类型复杂、多样,明确其定义和分类,有利于优化产品注册申报路径,加速各类产品技术指南的发布,推动产品快速研发上市,助力我国药品监管与国际接轨。本文通过调研美国食品药品监督管理局(FDA)、欧洲药品管理局(EMA)、日本药品医疗器械综合机构(PMDA)等药品监管机构在法规层面对先进治疗药品的定义及监管分类情况,结合我国产品申报现状和审评积累,提出了对先进治疗药品分类和描述的建议,为我国相关监管政策制定提供参考。
In recent years, the development and registration of advanced therapy medicinal product (ATMP) have shown explosive growth, leading the third industrial revolution of biomedicine. Different types of cell and gene therapy products, such as CAR-T cell therapy products and AAV gene therapy products, have been approved worldwide, offering effective treatments for relapsed and refractory diseases. ATMP are typically genetically modified and/or manufactured via in vitro manipulation of cells or tissues, including genetically modified cells, nucleic acids, viral vectors, and tissue engineering products. Due to the complexity and diversity of ATMP, clarifying their definition and classification is conducive to optimizing the drug registration path, accelerating the release of technical guidelines for various products, promoting the rapid development and marketing of ATMP, and fostering international regulatory convergence. This article investigates the definition and regulatory classification of ATMP in FDA, EMA, and PMDA. It proposes a description and classification of ATMP for drug regulation in China based on the current landscape of ATMP applications and review experiences, which could serve as a reference for formulating relevant policies and regulations.
关 键 词 / Key words
先进治疗药品;药品监管;法律法规;分类;细胞治疗药品;基因治疗药品
advanced therapy medicinal product; drug regulation; laws and regulations; classification; cell therapy medicinal products; gene therapy medicinal products
近年来, 以细胞和基因治疗产品为代表的先进治疗药品(advanced therapy medicinal product,ATMP), 为癌症、遗传病、罕见病等疑难疾病的治疗带来了新的契机和选择。临床高价值牵引先进治疗领域迅速发展,占据全球生物医药产业创新高地。全球先进治疗产业快速升温,行业投融资热度高涨。据统计,预计到2025 年,全球基因治疗市场规模将达到305.4 亿美元,我国市场规模将达到178.9 亿美元[1]。根据Citeline 数据库统计,截至2024 年4 月,全球共有100 余种基因、细胞与RNA 产品获得批准上市,超过3700 余种产品(其中包括约55% 的基因治疗产品,53% 的细胞治疗产品)处于临床前或临床(约占30%)开发阶段[2]。虽然我国先进治疗产业起步较晚,但目前已发展成为全球细胞疗法研发热度最高的地区。根据ClinicalTrials.gov 网站不完全统计数据,我国细胞治疗临床试验数量及申报产品数量位居全球第二位,仅次于美国[3]。自2021年我国第一个嵌合抗原受体T 细胞(chimeric antigen receptor T cell,CAR-T)治疗药品获批上市以来,我国上市CAR-T 产品数量已占全球同类产品50% 以上,我国先进治疗产业的发展已迈入与国际先进水平“并跑”的新阶段。
政策支持方面,各国家和地区均在持续加大研发投入及政策支持,以期推动更多的ATMP 上市,同时催生不断突破的新兴技术用于ATMP 的开发。放眼国际,美国陆续发布《国家生物技术和生物制造法案》《美国生物技术与制造目标》,将细胞和基因治疗产品提升至国家生物制造战略高度[4]。欧盟委员会(European Commission,EC)2020 年底发布《欧洲药品战略》,并致力于推动欧洲药品立法改革,预期将显著影响未来先进治疗领域的市场格局[5]。纵观国内,国家发展和改革委员会先后发布《“十四五”生物经济发展规划》《产业结构调整指导目录(2024 年本)》等,重点加强细胞和基因治疗产品等新兴领域的前瞻性布局,大力推动相关行业高质量发展和产业结构转型升级。各省市地区陆续出台相关政策规章,持续加力支持细胞与基因治疗等领域科技创新与产业发展。
国际合作与协调方面,根据《鼓励外商投资产业目录(2022年版)》,细胞治疗药物研发与生产(禁止外商投资领域除外)已被纳入鼓励范畴[6]。此外,2023年8 月,国务院印发《关于进一步优化外商投资环境 加大吸引外商投资力度的意见》,提出:“鼓励外商投资企业依法在境内开展境外已上市细胞和基因治疗药品临床试验”[7]。随着各项鼓励政策逐步放宽对细胞与基因治疗技术的市场准入,未来ATMP 研发国际合作将逐步深化,技术流动日益频繁,全球竞争日趋加剧,对国际标准协调及监管趋同提出了更高的要求。
全球诸多国家和地区不断致力于强化ATMP 的监管体系顶层设计和监管能力建设。美国食品药品监督管理局(Food and Drug Administration,FDA)、欧洲药品管理局(European Medicines Agency,EMA)、日本药品医疗器械综合机构(Pharmaceutical and Medical Device Agency,PMDA)等药品监管机构逐步建立了ATMP 的监管框架,积极制定发布相关法规及指南,并不断完善[8]。据不完全统计,全球各监管机构迄今已发布400 余个细胞与基因治疗产品相关的技术指南。部分国家和地区在法律法规层面明确了这类药品的定义及分类,并制定激励政策(如特殊审评程序等)加速产品审批上市。中国国家药品监督管理局(National Medical Products Administration,NMPA)目前已发布这类产品相关技术指导原则30 余个,覆盖研发、注册、工业化生产、上市后变更等各个阶段。在我国,该类产品适用于鼓励创新、加速审评审批等多种程序,但目前尚未在法律法规层面明确该类药品的分类与定义,其名称和分类尚未形成行业共识,名称使用及分类的不统一,不利于规范监管、行业沟通及国际协调。分类与产业发展紧密相关,明确ATMP 在我国的分类及定义迫在眉睫。
针对此现状,本文基于对全球主要监管机构ATMP 监管框架及分类与定义科学体系的调研和梳理分析,结合我国先进治疗产业发展现状、药品监管现状、国际对话与协调需求以及定义与分类专家研讨会共识,提出ATMP分类与定义的主要考虑和建议,以统一规范我国对这类药品的用词用语, 为后续进一步优化ATMP 监管体系提供参考,助力监管与国际接轨且标准协调一致,从而加速推进ATMP 的研发申报及审批上市。
1、国际ATMP 监管现状
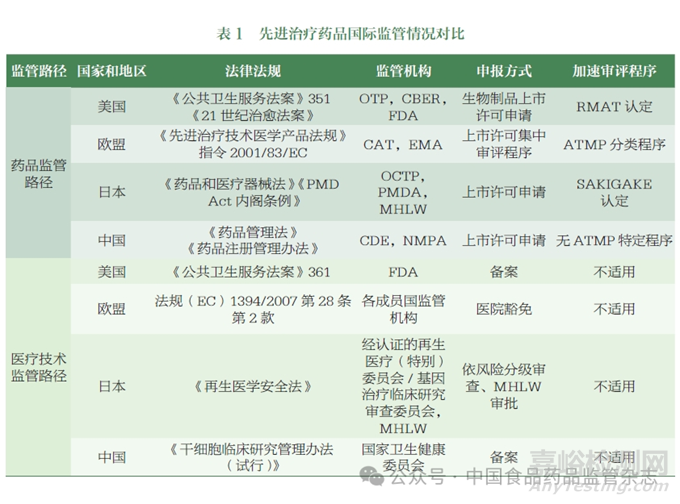
美国、欧盟和日本等国家和地区对先进疗法均存在药品监管和医疗技术监管两条路径,其中日本、中国涉及两个机构分别负责药品和医疗技术的监管。FDA、EMA、PMDA 等药品监管机构在法律法规层面均明确了该类产品的定义和分类, 并为该类产品专门设置了专业审评办公室。世界卫生组织(World Health Organization,WHO) 于2023年发布了ATMP 的定义和分类相关的技术指南。为加速具有临床价值的产品快速审批上市,FDA、PMDA 均对该类药品出台了加速审评程序,EMA 为该类产品审评审批成立了专门的先进治疗委员会(Committee for Advanced Therapies,CAT),并特别设置了ATMP 分类程序。
(一)美国FDA 监管概况
1. 分类和释义
美国FDA 在法规层面对该类产品拟定了3 种名称,包括人体细胞组织产品(human cells, tissues, or cellular or tissue-based products,HCT/Ps)、再生医学产品(regenerative medicine therapy,RMT)、细胞和基因治疗产品(cell and gene therapy, CGT)。3 类产品的释义和分类情况如下。
人体细胞组织产品(HCT/Ps):《公共卫生服务法案》(Public Health Service Act, PHS Act)对HCT/Ps 按照体外操作程度和使用情形进行风险评估,分为两类:PHS 361 产品和PHS 351 产品[9]。PHS 361 产品只需要在FDA 对其机构和产品进行登记,不需要申报上市;由于PHS 351 产品风险高, 除需进行机构和产品登记外,还需向FDA 提交新药临床试验申请(investigational new drug,IND) 和生物制品上市许可申请(biologics license application,BLA)。《联邦法规汇编》(Code of Federal Regulations)第21 章第1271 部分是细胞治疗产品审批主要的参考法规。该法规明确HCT/Ps 是指含有人体细胞或组织,或者由人体细胞或组织构成,并用于植入、移植、输注或转移至人类受者体内的物质[10]。
再生医学产品(RMT):《21世纪治愈法案》(21st Century Cures)Sec.3033 定义,再生医学产品包括细胞产品、组织工程产品、组合产品和部分基因修饰细胞产品[11]。
细胞和基因治疗产品(CGT):FDA 于1993 年发布了《联邦公报》(Federal Register), 显示细胞和基因治疗产品分为体细胞治疗产品、基因治疗产品、用于生产体细胞治疗产品的辅助产品、组合产品[12]。
2. 审评部门及鼓励政策
细胞、组织与基因治疗产品审评和批准具体由FDA 生物制品审评与研究中心(Center for Biologics Evaluation and Research,CBER)下辖的治疗性产品办公室(Office of Therapeutic Products,OTP)负责。OTP 由6 个办公室组成,包括3 个药学(Chemistry, Manufacturing,Control, CMC)审评办公室和其他3 个审评办公室,分别为:基因治疗CMC 办公室、细胞治疗和人体组织CMC 办公室、血浆蛋白治疗学CMC 办公室、临床评估办公室、药理毒理学办公室、审评管理办公室,OTP 拥有300 余名审评专家[13]。
《21 世纪治愈法案》明确,在IND 申报时或Ⅱ 期临床试验完成前可申请再生医学先进疗法(Regenerative Medicine Advanced Therapy,RMAT)认定,认定后将带来快速通道、突破性疗法的所有优惠政策,包括与FDA 开展早期沟通交流、加速批准等[14]。
为提高ATMP 的监管水平,FDA CBER 成立了先进技术团队(CBER Advanced Technology Team,CATT)[15]。考虑到新技术对产品研发生产和质量控制较为重要,监管方面经验有限,CBER 专门设置了CATT 会议针对该新技术进行讨论,拟通过此类会议完善对新技术监管方法和路径。2021 年,OTP 开始举办系列培训(OTP AMAT Training Program),涉及学术报告、课程学习、生产实践和培训会议等不同形式,为FDA 审评人员提供学习该领域前沿知识的机会,从而加深对细胞和基因治疗先进技术的理解[16]。
(二)欧盟EMA 监管概况
欧盟地区ATMP 整体监管体系由法律、法规、法令、指南4 级框架构成,其中法规(EC)No 1394/2007《先进治疗技术医学产品法规》为针对ATMP的专门法规[17], 明确了ATMP监管和上市程序的总体框架:ATMP 的上市许可由EMA 进行集中审评审批,由EMA/EU 集中授权,批准在所有欧盟成员国上市;各成员国可依据“医疗豁免”条款及各国家和地区医疗法规批准ATMP 在医疗机构内部使用。在指令2001/83/EC[18] 及法规(EC)No 1394/2007 对ATMP的分类及定义作了法规层面的界定和释义。同时,相继发布相关文件及程序,支持科学分类及产品加速审评审批。
1. 分类和释义
根据法规(EC)No 1394/2007,ATMP 总体划分为4 类,包括细胞治疗产品(somatic cell therapy medicinal product,SCTMP)、基因治疗产品(gene therapy medicinal product,GTMP)、组织工程产品(tissue engineered product,TEP)、组合产品(combined advanced therapy medicinal product,cATMP)。
细胞治疗产品(SCTMP):包含或由细胞或组织,经过实质操作使其与预期临床用途相关的生物学特性、生理功能或结构特性被改变或使其在受体和供体中表现不同的基本功能。通过细胞或组织的药理学、免疫学或代谢作用,具有用于人体的特性,用于预防、治疗或诊断疾病。
基因治疗产品(GTMP):产品的活性组分包含或由重组核酸组成,用于人体以调节、修复、替换、增加或删减一段基因序列。其治疗、预防或诊断作用与之所含的重组核酸序列或该序列的基因表达产物直接相关。GTMP 不包括抗感染性疾病的疫苗。
组织工程产品(TEP):包含或由人类或动物来源的工程细胞或组织,可用于再生、修复或替换人的组织。工程细胞或组织经过实质性操作使其与预期用途相关的生物学特性、生理功能或结构特性被改变或使其在供者和受体中表现不同的基本功能。包括含有或由活性或非活性的细胞或组织组成的产品。含有或完全由非活性细胞或组织组成的且不通过药理学、免疫学或代谢作用发挥效用的产品除外。
组合产品(cATMP):包含一种或多种医疗器械或活性可植入性医疗器械(如生物材料、支架、基质等)作为药物的组成部分。其细胞或组织部分必须包含活的细胞或组织,或者包含非活性细胞或组织,但其相对于器械部分发挥对人体的主要功能。
虽然欧盟在法律法规层面提出ATMP 的总体分类框架及各类产品的释义,但鉴于此类产品的复杂性和创新性,EMA 还针对ATMP 建立了较完整且精细化的科学分类支持系统,包括设置特定分类界定程序、发布分类建议书[19] 及分类界定清单。在EMA 进行正式上市审评前由申请人提交产品的分类界定申请,由CAT 于60 个工作日时限内给出产品分类的科学建议。EMA 发布了分类建议书,以决策树思维导图的形式提供了具体分类的科学依据及判定逻辑,并明确存在交叉属性的类别归属优先级,EMA强调出于安全的考虑,对于可能属于GTMP、SCTMP 及TEP定义范围内的产品应优先被视为GTMP, 同时符合SCTMP及TEP 定义的产品优先被视为TEP[20]。例如工程化CAR-T,其作用机制是通过修改细胞中的基因来实现治疗效果,其在欧盟归类于GTMP。此外,EMA 定期发布不断更新的ATMP 科学分类推荐清单,提供产品分类界定实例作为后续参考。
2. 审评部门及鼓励政策
在欧盟由EMA 负责ATMP药物的集中程序审评。EMA 为ATMP 的审评专门设立了CAT,主要负责ATMP 的质量、安全性及有效性技术审评、分类界定、相关科学支持等。CAT 为多学科委员会,由60 余名审评专家组成,人员包括主席/ 副主席、人用药品委员会(Committee for Medicinal Products for Human Use,CHMP)委员、欧盟委员会代表、欧盟各成员国代表、欧盟理事会患者组织代表及临床医生代表,其成员组成3 年更新一次[21]。CAT 审评后交由CHMP作出批准上市许可的推荐,最终由EMA 批准后颁发认证证书,由欧盟理事会做出最终对所有成员国均具有约束力的决定。
多种加快批准途径可适用于ATMP 的审评, 如优先药物快速审评(priority medicines,PRIME)、加速审评(accelerated assessment,AA)、附条件上市批准(conditional marketing authorization,CMA)、特殊情况下许可授权(authorization under exceptional circumstances, AEC) 等[22]。此外,ATMP 也适用于罕见病药物的认定等利好政策。ATMP 在欧盟集中审批的期限为210 个评估日, 加速审评程序通常可缩短至150 个评估日。
EMA 为支持ATMP 的开发,还提供以下激励政策:如在产品申报前通过创新工作组简报会议为新兴疗法和技术提供科学咨询和方案协助,联合卫生技术评估研究所提供科学建议等;通过数据共享计划加速阿尔茨海默病治疗药物开发;微、小、中型企业质量和非临床数据的认证以及系列减费措施等。
(三)日本PMDA 监管概况
1. 分类和释义
对于经过注册临床研究及上市申请的商业化再生医学产品,日本在各级法律法规、条例、指南性文件中均有相关要求及进一步阐释。
在法律法规层面,《医药品、医疗器械的品质、功效及安全性保证等有关法律》(通常称《药品和医疗器械法》,PMD Act)中对“再生医学产品”的定义如下:①人类或动物细胞经培养或其他处理后获得的产品,可用于人类或动物医疗保健,其功能包括:对人类或动物身体进行结构或功能的重建、修复;治疗、预防人体或动物的疾病。②可导入人类或动物细胞,并含有可在其体内表达的基因,以达到治疗人类或动物疾病的目的[23]。在《PMDAct 内阁条例》中规定了以下3类产品为再生医学产品:①加工的人类细胞产品,如诱导多能干细胞衍生的产品、人胚干细胞衍生的产品或体细胞产品。②加工的动物细胞产品。③基因治疗产品[24]。
在指南层面,《关于确保加工细胞/ 组织来源产品的质量和安全性》指南中对细胞/ 组织加工产品定义如下:以治疗疾病、修复或重建组织为目的,对细胞/ 组织进行的:①人工增殖;②药物或化学药剂处理,以活化细胞/ 组织;③生物学特性改变;④与非细胞/组织成分的组合;⑤基因工程操作等。并且明确指出,组织的分离、组织的破碎、细胞的分离、特定细胞的分离、抗生素的处理、洗涤、γ 射线或其他手段的消毒、冷冻、解冻等都不被视为加工,但非同源细胞产品除外[25]。《关于确保基因治疗产品质量和安全性》指南中的基因治疗产品,是指再生医学产品中的体内基因治疗产品和体外基因修饰人体细胞治疗产品[26]。
2. 审评部门及鼓励政策
再生医学产品最终上市许可的批准和许可证的颁发由日本厚生劳动省(Ministry of Health, Labour and Welfare, MHLW)负责, 生产和上市申请的审评由独立行政法人PMDA 下辖的细胞与组织产品办公室(Office of Cellular and Tissue-based Products, OCTP) 负责, 核查由PMDA 下辖的生产质量和合规性办公室负责,检验由日本国立医药品食品卫生研究所的细胞治疗产品部负责。PMDA 医疗器械审查部门的再生医疗制品审查组、生物源器械办公室负责再生医学药械组合产品审查。
在日本首先或同时进行申请的创新再生医学产品, 如基于非临床研究和早期临床试验的结果预期显著有效, 可获得SAKIGAKE 认定,认定后将带来优先咨询、优先审查等优惠政策,结合附条件和有时间限制的批准途径,可快速推进再生医学产品进入市场[27]。
(四)WHO 监管进展
WHO 于2020 年发布相关技术文件,阐述了细胞治疗产品的国际非专利药名的制定考虑[28]。2023 发布技术文件, 明确ATMP 是指任何经复杂体外操作和(或)进行非同源使用的细胞、基因治疗产品或组织工程产品[29]。ATMP 通常采用体细胞或组织经基因修饰和(或)体外操作制备,产品种类包括核酸、病毒载体和非病毒载体、重组细菌细胞和重组溶瘤病毒等。异种来源细胞或组织属于ATMP 范畴, 但考虑其复杂性不纳入文件讨论范围内。该文件明确将ATMP 分为细胞治疗产品、基因治疗产品、组织工程产品和组合产品,并在名词解释部分明确了各类产品的释义。
细胞治疗产品:由人体有核细胞组成的细胞产品,用于体内细胞替换和组织重建,和(或)通过调节该细胞的药理、免疫和代谢状态进而治疗或预防人体疾病的产品。
基因治疗产品:含有核酸(质粒、mRNA 或DNA) 的药品,通过调节、修复、替换、增加或删除特定基因序列发挥预期治疗效果,基因治疗产品包括非病毒载体、病毒载体和基因修饰细胞,也包括非重组溶瘤病毒产品。
组织工程产品:经过体外复杂操作和(或)非同源使用的由人体有核细胞组成的药品,用于组织修复、替换、重建或再生。
组合产品:ATMP 包括由医疗器械、支架、基质等各部分组成一个整体给药,其中器械或支持性结构有助于整体产品发挥治疗作用。
2、国际ATMP 监管对比分析及对我国的启示
(一)我国ATMP 监管现状
目前,我国对于细胞治疗产品采用双轨制进行管理,包括药品路径向NMPA 申报,按照生物制品审评审批上市,或由国家卫生健康委员会对医疗技术施行备案管理。目前我国药品监管机构尚未在法规层面拟定该类药品的定义和分类。2017 年, 原国家食品药品监督管理总局发布了《细胞治疗产品研究与评价技术指导原则(试行)》,该文件中细胞治疗产品是指用于治疗人的疾病,来源、操作和临床试验过程符合伦理要求,按照药品管理相关法规进行研发和注册申报的人体来源的活细胞产品[30]。根据2020 年发布的《生物制品注册分类及申报资料要求》,细胞治疗和基因治疗产品属于治疗用生物制品中的一类[31]。另外,《中国药典》2020 年版收录的“人用基因治疗制品总论”对人用基因治疗制品的定义进行了描述:通常由含有工程化基因构建体的载体或递送系统组成,其活性成分可为DNA、RNA、基因改造的病毒、细菌或细胞,通过将外源基因导入靶细胞或组织,替代、补偿、阻断、修正特定基因,以达到治疗疾病的目的[32]。
技术指南方面,NMPA 已制定发布的ATMP 技术指南30 余个,其他可以适用的通用型指南约100 余个,覆盖药学、非临床和临床研究领域,产品类型包括免疫细胞治疗、体内基因治疗、基因修饰细胞、人源干细胞、溶瘤病毒等。目前我国尚未在法规层面拟定ATMP 的范围、分类和定义,尚无法规和程序解释方面的技术文件对其进行清晰地分类和界定。
(二)国内外ATMP 监管对比分析
国内外ATMP 监管情况见表1。各国家和地区均存在药品监管和医疗技术监管两条路径,其中日本和中国涉及两个机构分别负责药品和医疗技术的监管。政策法规方面,美欧日及我国均出台了相应的法律法规及指导原则,其中美欧日在高等级的法律文件中对ATMP 的定义和分类进行了明确,我国则主要以指导性文件为主。监管主体方面,美欧日均有单独设立的ATMP 审评部门,如OTP(美国)、CAT(欧盟)、OCTP(日本)等,我国由药品审评中心(Center for Drug Evaluation,CDE)下设的生物制品药学部、药理毒理学部、生物制品临床部等部门联合审评完成,暂未组建单独的审评部门。认定程序方面,美欧日均设立有针对ATMP 的认定程序, 包括RMAT 认定( 美国)、ATMP 分类程序(欧盟)、SAKIGAKE 认定(日本),经认定后,可以按照加速/ 附条件审评路径申报注册上市[33], 我国暂未有对ATMP的特殊认定程序。医疗技术方面,除欧盟外(主管当局批准获得“医院豁免”),其余各国家和地区均以备案形式进行监管[34]。
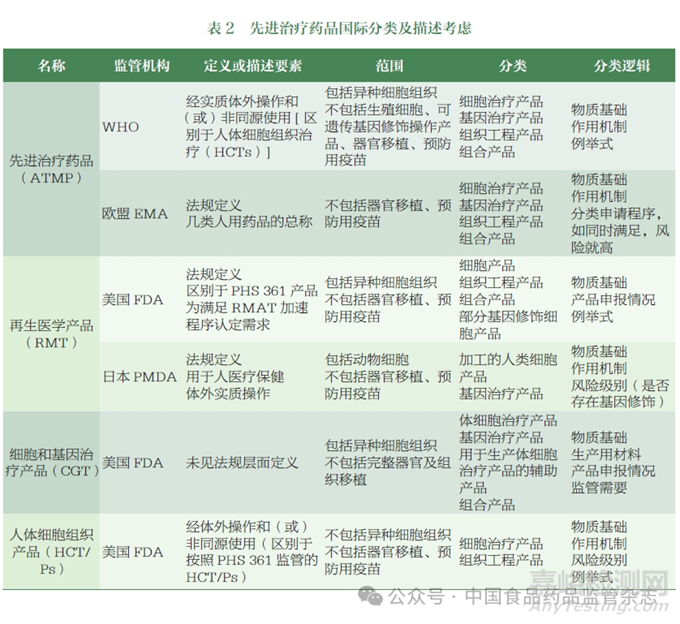
各监管机构基于不同的物质基础、作用机制以及监管需要等因素,对ATMP 进行了分类(表2)。WHO 明确ATMP 需经实质体外操作和(或)非同源使用的产品,与EMA 的分类和适用范围相似,包括细胞治疗产品、基因治疗产品、组织工程产品和组合产品,均不包括器官移植和预防用疫苗。FDA 对于ATMP 无统一、明确分类, 基于监管需求,将满足RMAT 加速程序认定的细胞、组织工程等产品定义为再生医学产品(RMT)。除此之外,FDA 还将用于生产细胞治疗产品的辅助产品纳入了细胞和基因治疗产品(CGT)中。PMDA统一定义为再生医学产品,明确该类产品需经体外实质操作,与WHO 类似,分类包括细胞和基因治疗产品。各类产品的描述方面,国际监管机构采用名词解释、描述和例举等方式阐明不同类别药品的定义或描述。
综上,国际先进监管机构在法律法规层面明确了ATMP 的定义及分类,并在科学层面结合产品的物质基础、体外操作特点、功能用途等方面,采用描述、例举等方式明确了各类产品的释义, 为该类产品审评技术标准体系建设和加速审批制度建立奠定了良好监管基础。现阶段我国尚未在药品监管法规层面明确其范围和分类, 也未针对该类产品设置特殊认定程序和与之相应的加速审评审批机制。针对这类产品制定明确的分类与界定,有利于制定相应的技术要求服务于产品的研究与开发,同时, 也有利于制定相关的监管政策与鼓励措施以促进产业的发展。
3、我国ATMP 分类和描述的建议
目前国内外ATMP 研发呈现类型多样化、工艺路线差异化、产品设计创新化等变化趋势。结合国外监管机构ATMP 监管分类的调研情况,CDE 组织相关部门梳理了国内相关产品的申报情况,初步提出了我国ATMP 的分类和描述,召开了专家研讨会,邀请学术界、工业界和监管领域的专家围绕名称拟定、分类和描述考虑要点开展了讨论,形成了以下建议。
(一)名称和描述
参考FDA、EMA、PMDA及WHO 等监管机构命名,结合国内现阶段工业界所用术语,该类产品的名称拟定时应主要考虑与国际接轨,同时体现产品的创新性和技术先进性,从而鼓励国内相关行业发展,并前瞻性地为未来新兴技术药品预留监管空间,可参考WHO 和EMA 将总的名称与具体分类结合考虑,确保总的名称突出药品属性,覆盖绝大多数相关药品种类,同时做好细化分类的归属。考虑到“细胞和基因治疗产品”主要体现产品物质基础,“再生医学产品”主要体现产品功能,相对局限,“先进治疗药品”的覆盖范围相对较广,并与WHO 等国际监管机构协调一致,建议采用“先进治疗药品”作为这类产品的中文名称,英文名称为“ATMP”。
参考国际监管机构对该类产品的释义,结合专家意见,笔者认为“先进治疗药品”的描述中应考虑纳入物质基础、工艺特点、功能用途等,同时基于体外操作程度和使用情形等对医疗技术相关边界产品的分类进行界定。考虑到该类产品按照药品监管,笔者将“体外操作”和“功能用途”作为ATMP 的描述要素, 并根据我国相关产品申报现状和未来发展趋势,基于活性成分的多样性及创新性完善相关描述。即描述为:“先进治疗药品是指经体外操作生产并在体内发挥作用的细胞治疗药品、基因治疗药品或组织工程药品,以及采用其他先进技术/ 方法生产的创新型药品等。”ATMP 的生产和研究过程需符合我国伦理方面的要求。输血用的血液成分、移植用的造血干细胞等不属于ATMP 的范畴。
物质基础方面,可考虑纳入目前国内申报量较大的产品类型如细胞治疗产品、基因治疗产品,同时纳入组织工程产品和其他新兴技术产品, 为新技术预留接口。工艺特点方面,结合FDA、EMA、WHO 等相关定义,通过体外简单操作制备且同源使用的细胞组织治疗产品不属于ATMP。因此笔者参考国际监管实践,结合产品生产常见工艺步骤,对描述中的“体外操作”明确了简单的释义,主要包括分离、纯化、扩增、基因修饰、基因编辑等。后续如有需要,将参考FDA 和其他监管机构的做法,进一步明确“体外操作”“同源使用”等定义。功能用途方面,参考国内生物制品监管现状,考虑到目前申报的产品主要为治疗用生物制品,因此描述中仅明确为治疗药品,暂不涉及预防、诊断相关用途。
结合专家建议,笔者对于医疗技术、涉及使用生殖细胞的产品等边界产品进一步开展了分类与界定。对于医疗技术,描述中考虑明确排除归属国家卫生健康委员会管理的输血、器官/ 组织移植等医疗技术。需要说明的是,这里的移植是指医疗机构的治疗手段,而非给药途径。例如,移植给药的基因修饰造血干细胞,由于其体外进行了复杂生产操作,因此也属于ATMP 范畴。对于涉及使用生殖细胞的产品和一些可遗传的基因修饰操作,由于来源和操作有多种不同情形,因此描述中明确了需满足我国伦理方面的要求。
(二)分类建议
为提升监管的科学性和有效性,鼓励药品研发不断创新,结合国际分类情况、产品物质基础、监管风险级别和国内产品研发申报现状, 笔者初步开展了ATMP 的分类研究, 并针对类别划分合理性和相关产品归属等进行了讨论。
1. 类别划分
研发申报方面,现阶段CDE受理审评的ATMP 种类和数量逐年增多,2023 年ATMP 临床试验的申报量达到生物制品总临床试验申报量的13%。截至2024年3 月,NMPA 批准了5 个CAR-T 细胞治疗药品、2 个基因治疗药品上市。获得默示许可开展临床试验的产品中,活性成分主要包括免疫细胞、干细胞等细胞产品,腺相关病毒、腺病毒等携带转基因的病毒载体产品,具有选择性增殖和溶瘤活性的溶瘤病毒产品等[35]。近年来,个性化肿瘤新生抗原产品的数量开始增长。另外,还有个别组织工程产品、组合产品申报。
结合研发申报现状、国内外监管法规/ 指南制定情况,为提升审评审批和监管效率,建议将我国ATMP 分为细胞治疗药品和基因治疗药品,对于难以分入这两类的药品,可考虑暂时单独设置一个类别“其他”,待认知成熟后再独立成为一类或进一步细分。例如,对于EMA、WHO 分类中的组织工程产品与组合产品,我国目前研发申报数量较少,现阶段建议归入其他类。具体类别的排放顺序方面, 可参考国际监管机构对该类药品按照产品研发成熟度由高至低、风险等级由低至高的排列顺序, 因此进行如下排列。
第一类:细胞治疗药品。
第二类:基因治疗药品。
第三类:其他。
2. 细胞治疗药品描述、亚类
不同细胞治疗药品在细胞来源、类型以及产品的复杂性方面各有差异。例如,细胞可以是自我更新的干细胞、分化方向确定的祖细胞或发挥特定生理功能的终末分化的细胞[36]。细胞可以来源于自体,也可以来源于同种异体或异种。此外,细胞也可能是经过基因修饰的[37]。这些细胞可以单独使用,也可以与生物大分子、化学小分子或结构材料联合使用。作用机制方面,干细胞治疗药品可通过补充或替换患者的受损细胞发挥组织重建作用[38],CAR-T 等免疫细胞可通过特异性杀伤肿瘤细胞发挥其生物学作用。结合国际监管机构对细胞治疗药品的描述,细胞治疗药品亚类的划分,总体需要考虑物质基础(活性成分)、作用机制和功能用途,即建议划分为非基因修饰细胞治疗药品和基因修饰细胞治疗药品。
笔者结合国际监管机构对细胞治疗产品的释义, 考虑其药理、免疫、代谢调节和细胞替换组织重建两大功能进行了描述,明确其以治疗疾病为目的,并列举了具体亚类及常见例子。目前WHO 的释义中,细胞治疗药品的物质基础需为有核细胞,但考虑到目前我国已有正在开展临床试验的红细胞类、血小板类药品,因此不建议限制有核细胞。
基于物质基础,以细胞作为主要活性成分的药品,根据细胞治疗产品是否经基因修饰,科学层面可将其分为非基因修饰细胞产品和体外基因修饰细胞产品两个亚类。考虑到CAR-T 等基因修饰细胞一般通过CAR 基因表达结合靶细胞从而发挥主要肿瘤细胞杀伤作用,FDA、EMA、WHO 等监管机构一般将基因修饰细胞纳入基因治疗产品的范畴。专家讨论认为,细胞治疗产品未来发展需要做大量修饰(包括遗传修饰、生物材料的联合使用等),但其物质基础及特征与细胞类似,最终起作用的药物形式为细胞,国内行业传统称之为细胞治疗产品。因此虽然这类产品归类到细胞治疗产品与部分国际监管机构分类不同,但是结合我国细胞产品行业分类现状,为了便于我国监管机构管理,建议将基因修饰细胞纳入细胞治疗药品范畴。
对于诱导多能干细胞(induced pluripotent stem cell, iPSC)衍生细胞产品是否属于基因修饰细胞,需要基于终产品是否有符合基因修饰定义的操作来确定。专家讨论认为,仅限于重编程生产iPSC 种子细胞的基因操作不是基因修饰,如不涉及后续细胞药品的基因修饰,则属于非基因修饰细胞产品。
3. 基因治疗药品描述、亚类
早期的基因治疗药品主要通过引入功能性蛋白转基因序列发挥作用, 常见使用病毒载体和质粒DNA 载体。近年来较新的基因修饰工具层出不穷,包括microRNA 等直接作用的核酸序列、通过短发夹状RNA(shRNA)起作用的RNA 干扰(RNAi)、锌指核酸酶(ZFN)或转录激活因子样效应因子核酸酶(TALEN)、分子剪刀和CRISPR-Cas 等基因编辑方法。这些工具可能通过基因沉默、外显子跳跃、基因调节、基因敲低和核苷酸改变介导基因序列的修复、扩增或删除。最近全球范围内,一些携带转基因的细菌载体产品逐步开始临床前研发或临床试验,相关细菌来自乳球菌属、李斯特菌属和链球菌属等,进一步扩充了基因治疗药品的类型。
国际监管机构对基因治疗药品的描述中,例举了其活性成分,阐述了其作用机制和功能用途。其中WHO 和EMA 均明确, 基因治疗产品的治疗效果与其所包含的重组核酸序列或该序列的基因表达产物直接相关。笔者结合国际监管机构描述,建议在基因治疗产品的描述中强调特异性改变基因序列/表达、引入外源基因等相关作用机制,并例举常见的活性成分。结合目前国内研发申报的基因治疗产品类型,基于活性成分可将基因治疗产品进一步细分为:病毒载体类药品、核酸类药品(DNA、RNA 等)、基因编辑类药品(如CRISPR-Cas9等)、溶瘤微生物或其他新型微生物类产品。
2005 年,我国批准了经基因改造的溶瘤腺病毒产品H101 上市。溶瘤病毒通常能够特异性地在肿瘤细胞中复制、增殖,通过直接裂解或激活机体免疫反应杀伤肿瘤细胞[39]。目前在我国也有溶瘤细菌等产品正在开展临床试验,考虑到尽可能覆盖更广泛的产品种类,笔者采用“溶瘤微生物”统一描述包括溶瘤病毒、溶瘤细菌在内的产品。作用机制方面,非基因改造的溶瘤微生物类药品与传统意义上通过基因改造发挥作用的基因治疗药品不同。但如将其单独自成一类,其研发申报数量预计远少于细胞治疗药品。专家讨论认为,根据溶瘤病毒产品近年来的研究申报情况以及其发展趋势,溶瘤微生物类药品基本均需要进行基因修饰或操作,对于非基因修饰的溶瘤微生物产品,考虑到其药学、非临床和临床研究与基因治疗药品相似,且WHO 文件已明确将其纳入基因治疗产品中,为促进国际监管趋同,也考虑纳入基因治疗药品类别。近年来科学家开始探索一些新型微生物的临床疗效,例如噬菌体产品等。为了区分该类产品与肠道益生菌等微生物类药品,笔者采用“其他新型微生物”来表述这类产品。
4. 其他类ATMP
根据我国创新生物制品申报现状,目前存在一些产品申报量小、总体研发成熟度相对低,但考虑到新技术发展趋势预期今后可能形成一类,例如肿瘤新生抗原产品、细胞衍生物(如外泌体)等。因此笔者设置“其他”类将这些产品纳入ATMP 的范畴,从而鼓励创新型产品的研发申报。由于这一类产品创新性强,难以拟定适宜的文字描述,笔者通过例举亚类来表述。这类产品主要包括新型递送系统药品(如细胞载体)、个性化靶点生物治疗药品(如肿瘤新生抗原药品)、细胞衍生物药品(如外泌体)、具有药品属性的组织工程药品(如人工器官或组织)等。
考虑到基因治疗产品中已经包括基因递送工具相关产品,但还有很多递送系统将蛋白、小分子等递送进入细胞,笔者将这类除了递送基因以外的递送工具纳入,包括递送蛋白的脂质纳米粒、递送小分子的囊泡类结构载体等。细胞衍生物中的外泌体既可以作为载药工具,也可以作为活性成分,其可能包裹蛋白或基因。基于物质基础可将其进一步细分为天然外泌体药物、基因工程化外泌体药物、外泌体载药药品(小分子)。一些病毒、细菌等囊泡结构药物也可以由细胞分泌生产,如病毒样颗粒,这类产品目前申报较少,后续出现时可通过具体问题具体分析,考虑是否可以纳入细胞衍生物亚类。另外,目前大量申报的产品中有一类个性化肿瘤新生抗原产品,这类产品的活性成分可能包括基因修饰的细胞、DNA、RNA 甚至多肽。基于物质基础难以将这类产品纳入细胞或基因治疗药品,因此考虑放置于其他类。
现阶段细胞治疗药品主要以单一细胞类型为主要活性成分研发,逐渐有组合多种细胞类型的产品用于临床试验。我国已有肝脏类器官等产品已开始用于临床研究,脑、胃肠、胰腺类器官和人工角膜等一些组织工程产品也在迅速发展过程中。结合研发现状,笔者考虑将具有药品属性的人工组织、器官类药品纳入其他类。对于异种移植产品,国内尚未出现此类药品申报,专家建议目前暂不考虑纳入异种移植。结合国际发展现状,如果后续国内出现遗传修饰的动物组织或者器官产品申报,考虑到该类产品涉及动物检疫和公共卫生方面人畜共患病的防控,可能需要与其他监管机构联合监管。
5. 边界情形
在确认一个产品是否属于ATMP 和分类的实际操作过程中,可能存在多种复杂的情形。同一产品可根据物质基础和作用机制可能划分至不同类别中。例如,胰岛小体的细胞组成包括胰岛α细胞、胰岛β 细胞等多种细胞类型[40]。根据物质基础可以将其归属于细胞治疗药品[41],考虑到其基底膜完整也可以归属于其他类中的类器官/ 组织工程药品。另外,有些产品的作用机制目前尚不明确或存在多种机制,例如基因修饰的器官类产品,存在基因修饰相关的生物学活性和组织重建功能。该类产品也可能存在不同的分类方式(细胞治疗药品中的基因修饰细胞亚类,或组织工程药品),因此需要具体情况具体分析,在积累经验和调查研究的基础上,考虑根据风险和作用机制等制定进一步的类别划分原则。笔者期望在现有的讨论共识基础上,基于药品的特点开展更多、更细致的讨论和研究工作。
4、展 望
ATMP 是当前生物医药领域最具潜力和前景的发展方向,占据国际竞争、国家战略规划的重要板块。临床价值引领、全产业链政策支持、前沿技术创新驱动、不断的资本加持及科学严格审慎的监管护航,将共同推动我国先进治疗产业的高质量发展,培育生物医药的新质生产力,及时满足人民群众未被满足的临床需求。然而,需要认识到当前阶段ATMP 领域机遇与挑战并存,药物开发、行业发展、国际合作方面均面临诸多难点,全链条监管也面临严峻考验。在产品开发与产业方面,该类产品个性化程度高、创新性强,前沿技术与早期临床探索同步推进,技术迭代更新迅速,工艺复杂,专利壁垒较高,同时涉及人体遗传资源利用及伦理问题,研发难度大,投入研发成本较高、开发周期长,但成功率较低。当前阶段生物医药整体行业遭遇资本寒冬,产品管线推进受阻。目前上市产品数量有限,且产品定价高昂,医疗保险尚未全面覆盖,市场支付能力不足。在药物供给、可及性及可负担性方面尚存在较大缺口。因此,监管机构对于这类产品的研究和发展需要给予更多的关注与支持,提供更多清晰明确的监管政策和鼓励措施。
综上,笔者基于我国行业发展情况、国内监管现状及国际标准协调、专家研讨会共识综合考虑, 提出关于我国ATMP 分类与定义的建议,总体分类框架及逻辑与国际主要监管机构分类体系相接轨。同时,鉴于新兴技术发展的颠覆性及产品开发的不确定性,在分类中预留一定的空间。上述分类建议及主要考虑虽非法规层面的界定,期望通过分类体系的调研及建议,为后续监管分类相关的支持政策制定提供参考,并引导完善相应技术指南、标准体系及国际监管协调,从而进一步提升ATMP 的监管效能,加速相关产品的研发申报及审批上市,助力满足人民群众未被满足的临床需求。

来源:中国食品药品监管杂志


