您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-06-18 08:32
对于生物药来讲,给药方案或剂型的变更还是挺常见的,比如给药频率从Q2W调整为Q3W,给药剂量从mg/kg变为固定剂量,给药途径从静脉注射(IV)改为皮下注射(SC)。很多给药方案调整后,药物的暴露量(如AUC、Cmax)会随之出现变化,导致不能满足生物等效标准(80-125%),因此如何建立新的且与之前相似的药物暴露量-药效/安全性关系是面临的挑战之一。
从IV到SC,患者的依从性会得到很大改善。毕竟,IV通常需要在医疗机构用药,且输注时间久,输注相关不良反应多,SC无论在耐受性还是给药便利性方面都有其优势。过去传统的证明IV和SC给药途经的相似性是通过临床Ⅲ期有效性和安全性终点进行确证。随着时间的推移和技术的进步,已经有一些案例采用基于模型的PK终点和有效性/安全性数据结合的方式,并获得监管机构认可。因此,基于模型的药物开发(model-informed drug development, MIDD)使用逐渐增多。
本文的主题围绕IV/SC桥接研究展开。
关于生物药物皮下吸收过程、机制、影响因素等,之前已专门写过一篇,不再赘述,有兴趣的可自行翻阅。目前人体SC给药生物利用度和吸收速率的准确预测还存在诸多挑战。尚未发现可用于人体SC吸收预测的最佳临床前研究种属。比如小型猪与人体皮肤比较接近,但小型猪SC吸收速度比人体快5倍多。而采用灵长类动物数据进行预测则会过高估计人体生物利用度。体外系统也可用于辅助临床前SC吸收数据收集,但很难模拟SC吸收的复杂过程。体内各种蛋白间相互作用(与细胞表面靶点和可溶性靶点、FcRn、Fcγ受体的结合)以及由此产生的非线性药代动力学可能会进一步阻碍了对SC吸收机制的评估,并可能使得非房室方法不再可靠。另外,由于很多抗体呈浓度依赖性的清除,不同剂量间的清除率也可能会不同。MIDD对于理解SC吸收,进行种属间预测有重要价值。
在药物初次获批后,出于多种原因,经常需要引入替代剂量方案。但是,通过开展非劣效性临床试验以证明替代剂量方案与原始剂量方案等效,可能需要高昂的时间和成本,尤其是对于获批多种适应症的药物。这种情况下,基于模型的桥接可能提供了一种省时、省力、省预算,且监管可接受的路径。涉及到的模型主要定量药理模型,包括PK、PK-PD、暴露量-效应关系模型等。已经成功的案例有很多,比如三种已上市PD-1抗体nivolumab、pembrolizumab、avelumab将给药剂量从mg/kg方式调整为mg/人这一固定剂量方式。又如PD-L1抗体atezolizumab调整给药间隔,通过E-R分析,发现与已上市给药方案1200mg, Q3W相比,840mg, Q2W或1680mg, Q4W具备可比的安全性和有效性,支持剂量间互换,给予患者更灵活的用药选择。正是由于以上多个案例的成功,FDA出台了《Pharmacokinetic-Based Criteria for Supporting Alternative Dosing Regimens of Programmed Cell Death Receptor-1 (PD-1) or Programmed Cell Death-Ligand 1 (PD-L1) Blocking Antibodies for Treatment of Patients with Cancer》、EMA出台了《Model-based approaches for approval of alternative dosing regimens and routes of administration of (anti PD-1 and PD-L1) monoclonal antibodies》,以更好的指导企业开展基于模型的给药方案调整。
MIDD在IV/SC桥接研究中也有诸多成功案例。比如HER2抗体pertuzumab和trastuzumab皮下固定剂量给药组合方案的开发,包括群体PK、E-R分析等。 又如trastuzumab由IV改SC的群体PK和E-R分析。
通常来讲,需要证明新的治疗方案比现行方案的获益-风险相当或更优,至少要证明暴露量的等效性,或者证明暴露量有差异但不影响临床有效性或安全性。不过,由于不同给药方案可能对应不同的药时曲线,传统意义的等效性标准并不适用于桥接研究,通常以非劣效暴露进行评估。如果超出非劣的标准,则要通过E-R分析,判断这种差异是否对临床有效性、安全性产生影响。这一策略已经成功应用于已上市PD-1/L1抗体不同IV方案间的桥接,并被纳入FDA/EMA指导原则。
下表是目前为止FDA或文献公开的开展过IV/SC给药途径桥接研究的药物总结。其中,abatacept、belimumab、golimumab、tocilizumab这4个单药的IV和SC给药路径均已经获批。除了golimumab是先获批的SC,后获批IV,其它3个药物均是先IV后SC的顺序。此外还有4个产品,daratumumab、rituximab、trastuzumab、pertuzumab联用trastuzumab,最初以IV给药途径获批,后续以与透明质酸酶共制剂的方式获批的SC给药途径。为了满足第2种给药途径获批的要求,公开的做法包括证明比安慰剂更优,证明第2种给药途经的临床终点和PK终点(比如稳态谷浓度Ctrough)较第1种非劣。其中,以PK为终点的路径越来越受到重视。至于PK终点选择Cavg还是Ctrough,则需要依据非临床和早期临床数据而定,大部分已获批药物在两个终点都达到了非劣标准,FDA也建议最好两个终点都能达到非劣。不过,abatacept只选择了Ctrough作为PK终点,也成功获批,认为Cavg与该产品的药效无关,Ctrough更相关。所以,还是需要根据具体产品具体分析。
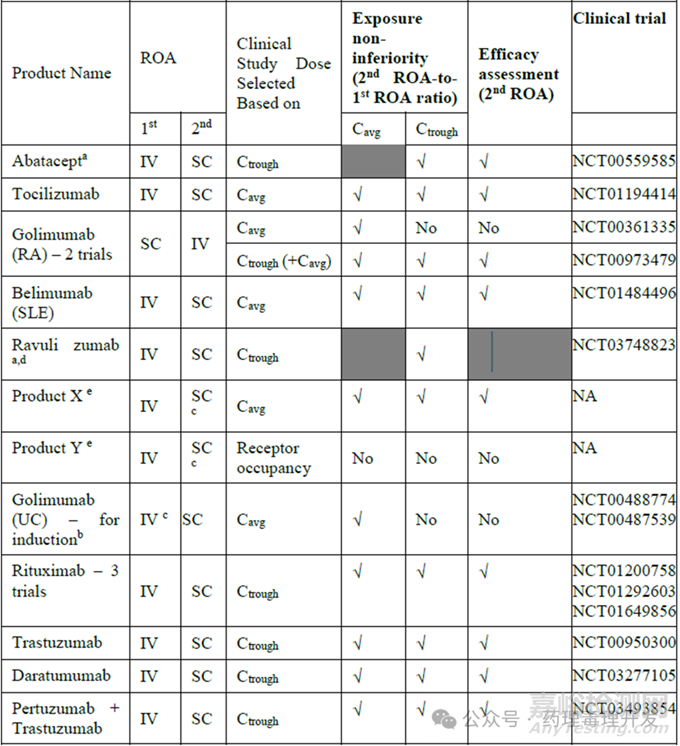
考虑到PK终点如此重要,FDA、EMA、PMDA、Health Canada对于不同给药途径桥接PK也给出很多建议:
1)不同给药途经之间的PK可比性:如果考虑采用模型进行桥接,需要对新给药途经的PK充分表征,并选择合适PK终点,建立不同给药途径之间的PK可比性标准。
2)E-R关系的良好表征:这点也容易理解,既然想通过PK的方式实现不同给药途径之间的桥接,那么暴露量与药物安全性、有效性之间的关系自然要研究清楚,这样PK可比才能与临床终点之间的可比建立联系。E-R分析还有助于PK终点选择、PK可比性标准确立。
3)PK终点的选择是基于E-R关系分析和对临床有效性、安全性的影响程度。
4)替代给药途经的剂量优化策略:如果选择基于模型、模拟的方式支持剂量选择,表征PK、E-R关系的方法需要state of the art,并向监管机构提供充分的、合理的说明。
那么IV-SC桥接的具体临床研究内容有哪些呢?
首先,获得IV给药的暴露量-效应数据。先上市给药途经是IV,首先要获得IV给药的暴露量与有效性、安全性之间的关系,并确定最佳的PK终点(比如Cavg或Ctrough)。
其次,获得SC给药的PK和生物利用度数据。这一步主要是开展SC给药的临床药理学研究。可以是多剂量、单次给药,伴随考察SC给药的安全性、耐受性数据。IV可以采用历史对照数据,也可以作为平行对照,在同一试验中考察。
然后,开展试验确认SC和IV给药之间的非劣性(PK和/或有效性终点),并提供SC给药的安全性、有效性等支持性证据。以Pertuzumab/Trastuzumab这对组合为例,早期上市的是IV给药治疗成人乳腺癌。为支持SC给药获批同样适应症,开展了一项开放、随机、非劣效设计的SC和IV对照的500例HER2阳性乳腺癌Ⅲ期临床研究。本试验的主要终点是PK指标Ctrough的非劣效,次要终点是伴随的安全性、有效性支持性证据。又比如Trastuzumab的IV-SC桥接案例,临床主要开展的HER2阳性乳腺癌患者中IV和SC对比的随机、开放Ⅲ期临床研究(596人),采用的共终点的设计,分别是PK指标Ctrough的非劣效、有效性指标pCR的非劣效。
但是,这一策略并不适用于所有IV-SC桥接情况,尤其是IV获批多个适应症,SC途径想桥接所有适应症的情况。以TNFα英夫利昔单抗为例,后续新增的SC途径想桥接之前IV获批的类风湿关节炎(RA)、强直性脊柱炎(AS)、克罗恩病(CD)、溃疡性结肠炎(UC)、银屑病(PsO)、银屑病关节炎(PsA)所有适应症。为支持这一目的,共开展了4项临床研究:1)RA患者中IV和SC对比的随机、双盲Ⅰ/Ⅲ期临床研究(407人),主要终点是药效的非劣;2)CD或UC患者中IV和SC对比的随机、开放Ⅰb期临床研究(181人),主要终点是PK指标Ctrough的非劣效;3)CD患者中SC和安慰剂对比的随机、双盲Ⅲ期临床研究(397人),主要终点是药效优效;4)UC患者中SC和安慰剂对比的随机、双盲Ⅲ期临床研究(548人),主要终点是药效优效。总体上还是桥接的思路,采用CD和UC两个适应症的PK、有效性终点,叠加RA适应症的有效性非劣设计,新的SC给药途径成功获批了所有5个适应症。只不过,这种临床设计策略还是有些复杂,涉及不同适应症的敏感性和代表性,比如药物在不同适应症之间的安全性表现不同,则不能进行简单的适应症之间的外推,需要结合产品特点、作用机制、前期IV给药临床数据等资料,并与监管机构充分沟通后确定。
此外,对于PK终点不能满足非劣的情况,自然需要开展SC给药的Ⅲ期有效性和安全性确证。对于IV给药已经获批A和B两个适应症,SC也已经获批A适应症,想扩增B适应症的情况,则仅需PK证据即可。
还有些情况很难获得PK稳态的实测数据,比如有些药物治疗方案是给药几次后,采用其它药物维持治疗。又如有些适应症(如肿瘤)患者的脱落率比较高。再如罕见病患者的入组极具挑战。对于获得PK稳态指标可行性不足的情况,可能需要用到MIDD,通过前期已有数据,对药物稳态时的Cavg或Ctrough等指标进行预测。对于采用MIDD的情况,需要对用于预测的数据、拟采用的模型进行充分论证。
最后,附上FDA和EMA批准的IV给药治疗性蛋白新增SC给药开展的临床研究内容。
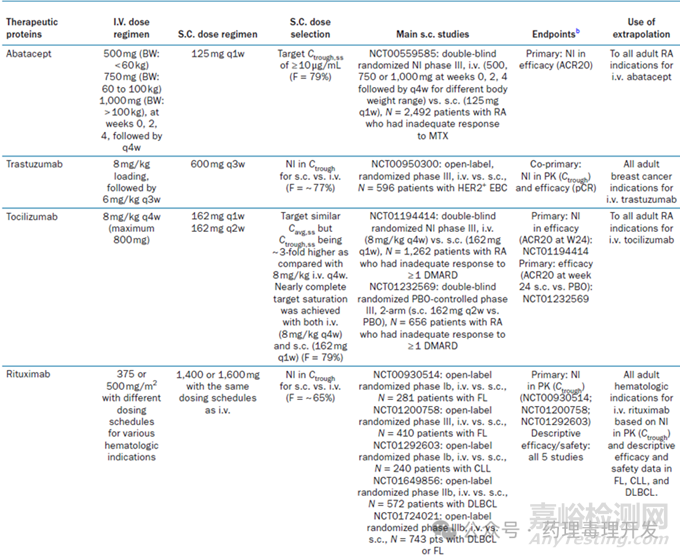
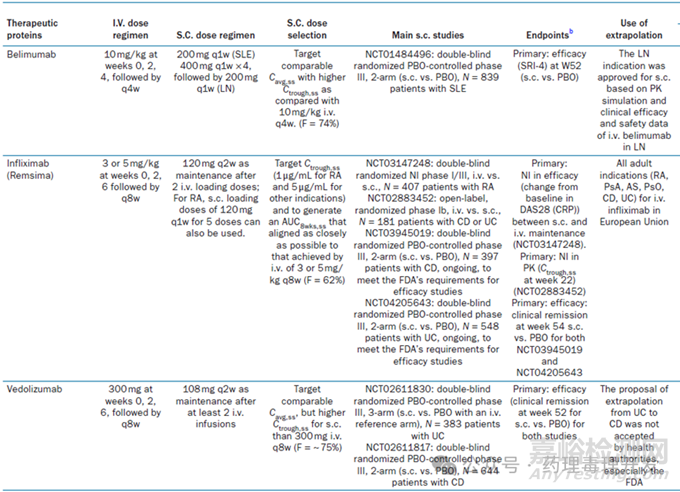

来源:药理毒理开发


