您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-06-20 08:47
摘要
生物制品的质量控制是其药学研究和评价的关键,贯穿药品的整个生命周期。随着生物制品相关分析技术的发展和上市后产品知识的积累,上市许可持有人(marketing authorization holder,MAH)可持续改进药物分析方法,增强质量风险管理。本文从监管角度出发,探讨关于治疗用生物制品分析方法变更的审评考量,分享分析方法变更的相关案例,为制药企业持续不断提升药物质量控制和完善分析方法变更申报资料提供参考。
关键词:治疗用生物制品;分析方法;全生命周期管理;上市后药学变更
生物制品的质量控制是其药学研究和评价的关键内容,贯穿药物的全生命周期管理。在开发期间研究者需要根据临床安全性、有效性、免疫原性和药动学影响对药物质量属性进行评估,以确定其是否为关键质量属性,针对关键质量属性建立相应的控制策略,选择特定检测项目的分析方法,并对检测结果制定合理的可接受范围[1-2]。分析方法作为质量控制的重要组成部分,决定了质量控制体系的可靠性和准确性。
分析方法的开发、验证、转移、变更和日常监测是连续且相互关联的,贯穿了产品生命周期管理的始终,不断推动着产品质量朝着更优的方向发展[3-4]。药物早期的研发、临床试验申请和注册过程往往经历分析方法的开发、建立、确认、验证、转移、再验证等过程,从而确保分析方法适用于药物的过程中控制、放行检测和稳定性检测[5]。在获批上市后,一方面上市许可持有人(marketing authorization holder,MAH)需要对生产工艺进行持续验证,不断积累商业化生产的研究数据;另一方面,随着更加先进和灵敏的分析技术的出现和成熟运用,MAH 利用这些新技术持续改进药物分析方法,采用更优的分析技术以增强产品质量风险管理能力[6]。应用新的质量控制分析方法,往往具有降低检验成本、提高方法稳健性、加快产品放行、减少实验动物使用等方面的优势。
本文将从监管角度出发,探讨生物制品分析方法变更的相关案例,为MAH 持续不断提升药品质量控制和完善上市后变更申报资料提供参考。
一、研究流程和一般策略
ICH Q8,Q9,Q10 和 Q11 等指导原则描述了用于药物开发的质量源于设计(quality by design,QbD)理念、方法和工具以及全生命周期的质量体系要求[6-8]。随着 QbD 在药物开发中的应用越来越广泛,已扩展到包括分析方法生命周期在内的分析方法开发和维护中。在分析质量源于设计(analyti-cal quality by design,AQbD)框架下,MAH 应充分利用专业知识和生产经验,积极运用风险评估和风险管理工具,开展持续的监测和验证,从而获得符合预期性能要求的质量可靠的分析方法[9]。基于科学的风险评估和全面的变更研究是分析方法生命周期内迭代更新的基础。
MAH 在进行分析方法的迭代更新研究时,可以参考相关的国内外技术指南,例如国家药品监督管理局的《已上市生物制品药学变更研究技术指导原则(试行)》、ICH Q12 的“药品生命周期管理的技术和监管考虑”、ICH Q14 的“分析方法开发(草案)等[10-11]。
生物制品分析方法的迭代变更与产品质量控制密切相关,由 MAH 对分析方法的变更进行风险评估,确定风险等级。MAH 可以参考《已上市生物制品药学变更研究技术指导原则(试行)》,同时结合分析方法特点和实际变更内容展开风险评估,根据风险等级执行变更研究,按照药品注册管理的规定及程序申报补充申请、备案或报告。变更执行后,MAH 仍需持续对分析方法的性能进行监测,收集产品质量相关信息。分析方法变更研究流程示例见图1。
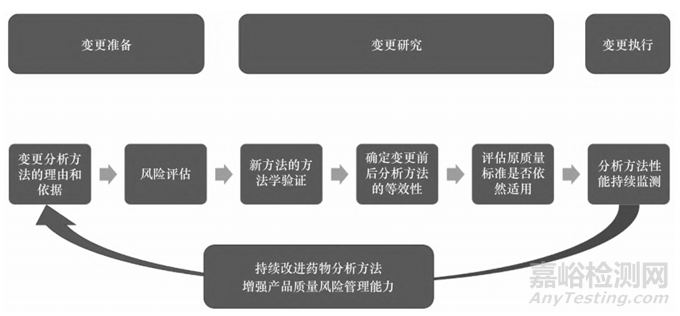
▲图1-分析方法变更研究流程示意图
1.1 变更理由和依据
分析方法在产品生命周期内的迭代更新应朝着更加准确、灵敏、稳健、快捷的目标努力,从而保证质量控制体系的先进性和可靠性。MAH应在申报资料中提供变更分析方法的理由和依据。通常导致分析方法更新的理由可能包括企业自身因素和外部因素。企业因素包括提高方法性能、降低检验成本、易于操作、加快产品放行、解决供应链问题等,或由于对产品的质量属性有更深人全面的认识而提出更多分析要求(如抗体中的痕量序列变异体[12])。外部因素包括《中华人民共和国药典》分析方法的升级或为了符合《中华人民共和国药典》各论要求、上市后批件遗留问题要求、生物类似药对应的参照药质量标准或分析方法升级、实验动物伦理压力等。
1.2 风险评估
MAH 应根据分析方法迭代更新的具体事项、可能后果和不确定性对预期变更进行风险评估,需要考虑的因素通常包括待检测质量属性的等级、技术复杂性和变更程度等。
1.2.1 质量属性等级
不同质量属性对于产品质量和疗效的影响类型和影响程度需要结合潜在临床影响(有效性、安全性、药动学和免疫原性)进行进一步风险评估。MAH 可以通过运用不同的风险评估工具,根据产品特定的研究、同类产品/平台经验外部文献知识确定质量属性等级。通常情况下,某一质量属性的等级越高,则用于控制该质量属性的分析方法变更的风险等级越高。
1.2.2 技术复杂性
基于动物、细胞和免疫化学的活性分析方法与理化分析方法相比,技术复杂性更高、潜在影响因素更多,因而变更风险等级更高。重组蛋白类产品的电荷变异体通常色谱图的组分峰复杂,某些峰间分离度较差,需要根据酸碱组分进行合并报告,风险等级较高。外观(颜色、澄清度)和鉴别项目属于定性的分析方法,如采用药典方法或成熟的技术时,与其他用于质量控制的定量分析方法相比技术复杂性较低。
1.2.3 变更程度
分析方法在整个生命周期内均可能发生变更,变更可能为同一分析方法内的参数微调和修改,或者完全替换成新的分析方法。耐用性研究中经验证的参数变更时,或仅在相同分析方法内进行一个或者多个参数的微调时,风险等级较低。药典方法是经过充分证明、技术成熟且广泛应用的分析方法,当将药典中同一检项下的一个检测方法变为另一个检测方法,或者由企业内部分析方法变为药典方法时,通常风险等级较低,但仍需要进行研究和评估。某一检测方法从药典方法变为非药典方法时,需要谨慎并进行全面合理的研究与评估。新技术更迭如果涉及分析方法原理变更,由于先验知识和使用经验的缺乏,在应用初期存在一些不确定性,风险等级较高。
1.3 变更研究策略
MAH 在确定分析方法变更的风险等级后,可参考相关法规和指导原则制定变更研究策略,开展相关研究。分析方法的迭代更新可以从以下方面开展研究:对新方法进行全面的方法学验证;采用变更前后的分析方法对代表性样品进行比对分析,从而确定变更前后分析方法的等效性;根据等效性研究结果评估原质量标准检测项目的可接受范围是否适用于更新的分析方法;如不再适用,需要重新拟定质量标准。质量标准的拟定可以参考相关文献、ICH Q6B 和《中华人民共和国药典》要求[1,6]。MAH 需要提供质量标准拟定依据及拟定过程,证明质量标准制定的合理性。ICH Q14“分析方法开发(草案)提出了一种分析方法变更评价的示例,见表1。
▲表1-ICH Q14 分析方法变更评价示例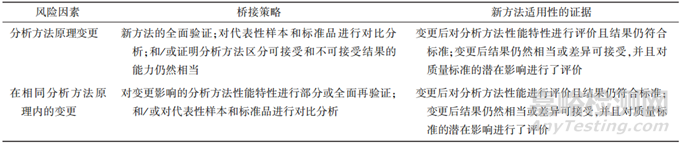
二、案例分析
2.1 纯度分析方法
生物制品具有复杂的高级结构、翻译后修饰和糖基化修饰,同时在贮存中容易发生聚集和降解,产生分子大小异构体和电荷异构体[13]。这些变体的存在可能对产品生物学活性、免疫原性及体内代谢产生影响,通常需要采用多种分析手段对生物制品的纯度进行控制。随着分析检测技术手段的升级,越来越多重现性好、灵敏度高、自动化程度高的分析方法用于替代传统分析方法,应用于药物的质量控制,例如采用毛细管凝胶电泳(capillary electrophore-sis-sodium dodecyl sulfate,CE-SDS)替代还原/非还原十二烷基硫酸钠-聚丙烯酰胺凝胶电泳( sodiumdodeeyl sulfate-polyacrylamide gel eleetrophoresis , SDS-PAGE)控制药物的分子大小异构体;采用成像毛细管等电聚焦(imaged capillary isoelectric focusing,iCIEF)毛细管等电聚焦电泳(capillary isoelectricfocusing,cIEF)替换定性的等电聚焦(isoelectric fo-cusing,EF)控制药物的电荷异构体[14-15]。
当与纯度相关的分析检测技术手段发生升级时,通常需要对新方法进行完整的方法学验证,同时对新旧方法的关键方法学性参数(如精密度、准确度、检测限)进行比较,新方法的灵敏度应不低于旧方法。在此案例中,某企业原本采用 IEF 凝胶分析联用考马斯亮蓝染色法进行原液和成品的放行与稳定性检测,以控制电荷异质性,但该产品电荷异质体组分复杂,不能准确定量;拟采用较新的cEF平台技术替代半定量IEF,以便更为准确地定量测量电荷异构体。研究者进行了方法学验证,包括专属性准确度、重复性、中间精密度、线性、范围、定量限、检测限、耐用性,并考察了方法的稳定性指示作用。cIEF 峰检测限为 0.3%,相比之下变更前 IEF 凝胶中条带的检测限为1%,拟定分析方法实现了定量检测,灵敏度更高。
研究者需要将现行方法与拟定方法比较,同步采用现行方法和拟定方法分析一定批次的代表性样品,从而确定变更前后分析方法的等效性。纯度和异构体检测项目通常具有稳定性指示意义,研究者可采用新旧分析方法对稳定性留样或强制破坏样品进行同步分析。此外,由于拟定分析方法可能对产品异构体的分离能力更好,研究者还应对新分析方法检测出的异构体组分峰或新的杂质峰进行分离鉴定和表征研究[16-17]。在此案例中,研究者分离不同的电荷异构体组分峰后,采用液质联用的方法表征蛋白质一级结构和糖类结构,对富集的电荷异构体进行结构表征和生物学活性检测,结果证明电荷异质性与C末端赖氨酸含量变化、天冬酰胺脱酰胺化和唾液酸修饰有关。
在此案例中,先前的原液和制剂质量标准限度范围不再适用于新的分析方法,研究者可根据代表性原液和制剂的放行数据、长期稳定性数据和分析方法的变异性拟定变更后检测方法的质量标准。研究者采用了68个原液批次(包括冻存留样和商业化批次)确立质量标准,分别计算了历史12个批次和当前 48 个批次的主峰、酸性峰和碱峰的均值±3SD 区间,拟定了原液放行质量标准范围。申请人应根据实际申报的具体情况(如质量属性特点、工艺能力、临床试验相关性等)选择合适的统计学方法。在制定成品的放行和货架期标准时还需要考虑制剂生产和贮存过程中发生的电荷异构体变化。审评机构将根据变更风险要求申请人基于更新的检定方法及质量标准与中国食品药品检定研究院接洽单项复核检定事宜。
综上,通过对代表性批次样品的分析和全面的方法学验证,研究者证明新方法的灵敏度和分离度有显著提高且具有稳定性指示意义,并对质量标准限度范围的适用性进行了重新评估,从而保证了质量控制体系的先进性和可靠性。
2.2 工艺相关杂质分析方法
生物制品的工艺相关杂质通常来源于细胞基质、细胞培养或下游工艺,包括宿主细胞蛋白( hostcell protein,HCP)、宿主细胞残留 DNA、细菌内毒素、色谱配基、工艺添加物等。工艺相关杂质应尽可能在下游纯化工艺中去除,以保障患者安全。风险等级较高的工艺相关杂质通常作为常规监测纳入原液放行质量标准。残留 HCP 具有潜在的免疫原性风险,还可导致蛋白聚集、降低最终产品的质量,影响制剂和处方的稳定性[18]。作为工艺相关杂质的控制重点,下文将对 HCP 残留量分析方法的变更研究进行探讨。
生物制品的 HCP 残留量通常采用酶联免疫吸附测定(enzyme-linked immunosorbent assay, ELISA)进行定量检测。ELISA 包被板中的多克隆抗HCP抗体决定了测定法的灵敏度和 HCP 覆盖率。在产品开发初期,研究者通常采用商业化试剂盒进行HCP 残留量的检测。进入关键性临床阶段以后,研究者将自行开发基于专属产品和特定工艺的ELISA分析方法。研究者可采用基于凝胶的分析方法(如2D-PAGE/2D-Western blot)或液质联用( liquid chro-matography-mass spectrometry,LC-MS)分析方法完成新旧方法 HCP 覆盖率的检测。关于拟定方法应达到的最小 HCP 覆盖率,目前监管机构尚未有明确共识,但新分析方法的 HCP 覆盖率理论上不应低于原分析方法。
例如,某企业申请采用新开发的多克隆抗CHOHCP 家免抗体代替原有的山羊抗体进行EISA 分析,从而实现更好的免疫应答。变更试剂盒抗体来源后,试剂盒对较低分子量和碱性残留蛋白的检出效率更高,采用 CHO HCP 标准品进行对比研究,原抗体检测覆盖率约68%,拟定方法抗体检测盖率约95%。申报资料对95个历史批次分别采用变更前后的检定方法检定 HCP,结果显示相同批次产品的 HCP 水平整体升高,超出原有质量标准限度和行业标准限度。本品仅进行了 HCP 试剂盒更新,生产工艺等其他方面均未发生变更,产品质量无实质变化,更新的检测试剂盒 HCP 覆盖率提高,申请人自检结果与中国食品药品检定研究院检定结果差异较小,基于充分的变更研究和风险评估,认为修订该技术标准具有较好的科学基础。需要关注的是,在分析方法变更的同时如伴随其他变更,不能判断产品质量是否发生变化时,放宽 HCP 杂质残留限度有可能增加患者的安全性风险,研究者需要结合人体暴露量、临床免疫原性和安全性数据、残留 HCP 的种类和风险等充分评估放宽 HCP质量标准限度的合理性。
当前生物制品残留 HCP含量的控制依然基于可检测的宿主残留蛋白总量。考虑到不同产品的纯化工艺对 HCP 的清除能力不同,且各种 HCP 的分子大小、结构、可检测性和安全性风险存在差异如何使新型 HCP 分析方法和控制手段应用于产品质量控制,实现更精准的基于风险的控制,有待监管机构和工业界共同探讨。近年来,基于LC/MS 的HCP 检测方法在鉴别和定量的性能上逐渐超越了常规免疫测定法。LC/MS分析方法可实现不同HCP的分离鉴定和分别定量,从而更好地针对其安全性风险实施控制,在考察下游纯化工艺对 HCP 的清除研究以及 HCP 覆盖率研究中都具有巨大的应用潜力[19-22] 。当 ELISA 方法检测到总 HCP 水平升高或其他异常的分析检测情况时,可采用替代方法对个别潜在高风险 HCP 进行针对性的监测。例如,某些特定 HCP 具有降解制剂中关键辅料聚山梨酯80的酶活性,从而影响制剂稳定性;可以开发基于酶活性的检测方法对特定 HCP 进行监测和控制。此外,研发机构在必要时可采用分离的高风险杂质开展进一步的免疫原性评估和毒理学研究,甚至从工程细胞构建的源头敲除高风险 HCP 相关基因。
2.3 活性分析方法
生物学活性是产品有效性的评估手段,是产品最关键的质量属性之一。生物制品的生物学活性测定应尽可能反映产品的作用机制。生物学活性的分析方法可用于定性鉴别,也可用于定量检测。结合不同制品的作用机制差异,研发机构可开发体内动物分析、细胞活性、结合活性等多种活性检测手段,目前胰岛素类产品已经可以用色谱法替代动物法但是一些激素类产品(如生长激素、促卵泡激素等)被《中华人民共和国药典》收录的生物学活性仍均基于实验动物,如小鼠、大鼠或家免等[23]。从体内生物测定法变更到体外生物测定法或理化分析法可减少使用实验室动物,符合国际实验动物 3R 原则即reduction(减少)、replacement(替代)、refinement(优化)[24-25]。研究者应说明用体外活性方法替代动物法的科学性和合理性,充分证明2种方法的相关性及体外活性测定方法的可行性。在具有充分依据的情况下,研究者可以申请用细胞活性分析方法替代基于动物的分析方法。
替代性的体外活性测定可以根据不同产品的作用机制开发,例如通过测试目的蛋白与膜受体结合的效率,或者通过测量目的蛋白刺激培养目标细胞内酶或辅助信使的能力来实现。报告基因法是待测生物制品的刺激反应元件或作用靶标和荧光素酶基因共同转染生物学活性测定细胞,当样品与细胞膜上的受体结合后,通过信号转导激活反应原件,导致荧光素酶的表达激活或抑制,从而在体外可使用光度计进行定量检测。某企业申请用葡萄糖-6-磷酸酶荧光素酶启动子系统代替家免实验检测胰岛素的生物学活性。研究者评估了方法的线性、重复性、中间精密度、准确度、范围、样品/标准溶液稳定性和专属性,结果满足验收标准。如果测得的胰岛素活性为不低于 15 U·mg-1,则认为按照美国药典(USPharmacopeia,USP)生物鉴别方法进行的检测符合要求。细胞受体检测方法通过与公司参考标准品比较测定了某个样品的相对效价百分比。研究者进行了方法学比对研究,结果表明在家免实验中生物鉴别呈阳性的9个批次(2个原料药生产厂生产的批次)在细胞受体检测中也呈阳性。监管方认为变更前后方法学比对研究的批次和数据有限,建议在初期采用2种分析方法并行进行原料药的生物活性分析,积累足够的放行检测数据并充分评估2种分析方法的相关性。此外,研究者需要关注变更后原质量标准是否依然适用。体内法通常为定性检测,而细胞分析方法作为可量化的指标,其标准限度应设定上限和下限。
2.4 快速微生物检测方法
无菌检测是生物制品放行检测中的常规安全性检测项目,通常根据《中华人民共和国药典》2020 年版通则 1101 无菌检查法检测,以确保产品无微生物污染。传统的微生物检测方法需要14d的培养时间,由分析人员目视检查。对于细胞治疗等短效期的产品和用于流行病防控的新型疫苗产品,长达2周的微生物检测时间可能成为制约产品放行和用于患者的限制因素。制药行业在过去多年间,不断致力于开发新型可替代的快速微生物检验技术。采用快速微生物检测技术替代传统微生物检测手段具有众多优势:可加快产品放行、降低生产成本,实现在线监控、保证数据完整性,同时对生产中的潜在污染更快响应。
目前应用较广泛的快速微生物检测技术主要基于培养微生物获取信号、直接测量微生物或分析细胞成分原理。常见的快速无菌检测系统有 MilliflexCelsis Advanc,BacT/Alert,Bactec 等。Celsis 检测系统和 Milliflex 检测系统均基于 ATP 生物发光成像系统,BacT/Alert 检测系统和 Bactec 检测系统基于对代谢产物CO,的检测。不同快速微生物检测系统的分析检测时间、测试能力、检测限存在差异[26-28]。
《中华人民共和国药典》、美国药典和欧洲药典均收录了微生物检验替代方法验证的相关指导原则,研究者可以根据微生物检验方法的定性或定量差异,开展替代方法的方法学验证。微生物检验替代方法的验证可参考《中华人民共和国药典》2020年版三部“药品微生物检验替代方法验证指导原则”开展。无菌检查法的替代验证属于定性检查应至少考察快速检验分析方法的专属性、检测限、重现性和耐用性。微生物限度检验的替代验证属于定量检查,应考察快速检验方法的准确度、精密度、专属性、定量限、线性、范围、重现性、耐用性。研究者还需要开展方法适用性研究,考察制剂是否存在生长抑制或产品干扰。研究者应证明替代方法与药典分析方法之间的等效性。等效性研究是一种比较试验,要求使用相同范围的微生物平行运行2 种方法。根据2种方法所产生数据的统计处理来证明替代方法结果等效或优于(不劣于)药典方法。如果替代方法产生的结果优于或等效药典方法,则认为替代无菌检查的性能等效。快速无菌检验替代方法的验证总结示例见表2。
▲表2-快速无菌检验替代方法的验证总结示例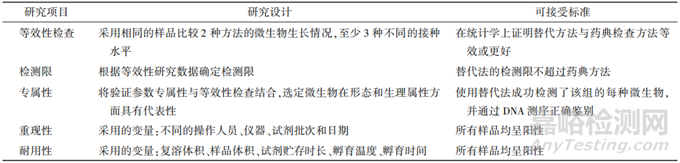
微生物检验替代方法的方法学验证和等效性研究所涉及菌株至少应包含微生物限度检查和无菌检查法规定的药典菌株,还应根据替代方法及样品特点增加相应的菌株。《中华人民共和国药典》中无菌检查法的实验菌株包括金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌、生孢梭菌、白念珠菌、黑曲霉;非无菌产品微生物限度检查(微生物计数法)的实验菌株包括金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌、白念珠菌、黑曲霉。此外,研究者可根据产品特点增加一些环境常见菌、生长缓慢并难以检测的菌株、对患者高风险的菌株和挑战微生物(如饥饿或应激菌株)。研究者应在申报资料中充分评估微生物种类和选择的合理性,例如,某公司在申请同步采用药典方法和替代方法用于产品放行的变更时,未纳人2种《中华人民共和国药典》2020 年版要求的实验菌株,审评机构建议其进行相关补充研究或就未纳人相关实验菌株的合理性予以解释。
考虑到传统的微生物检测方法已应用多年,监管方可能建议在应用替代分析方法的初期,同步采用药典分析方法和替代分析方法进行放行检测,从而积累足够批次的数据作为支持。当然,快速替代微生物检测方法也存在一些缺点,例如资金投入高单一供应商、难以检测生长缓慢的菌株或某些特定菌株等。为充分保障用药者的安全,建议申请人根据替代方法的局限性制订风险应对措施和质量控制策略。
三、讨论
分析方法的生命周期管理将在执行变更后持续进行[29]。研究者需要在日常运行中对更新的方法进行持续监测和定期评估,从而确认分析方法的性能符合预期,促进分析方法的持续改进[30]。持续性监测、工艺验证并积累商业化数据后,研究者还应对质量标准可接受范围的合理性进一步评估[31]。
分析方法在产品生命周期内的迭代更新应保证质量控制体系的先进性和可靠性[7]。MAH 应基于科学和风险的基础开展支持性研究,用充分的数据和逻辑支持变更。在分析方法发生变更的同时,如果生产物料和生产工艺没有发生变化,那么产品质量、安全性和有效性并不会发生实质性变化,更新后的分析方法将更有利于反映产品质量的真实状况,降低检测能力限制带来的不确定性,识别质量属性出现异常的情况。
平台分析方法的上市后变更面临着多个产品的同步实施。通常情况下,当待测样品的结构和属性相近的情况下,用于检测不同产品的平台方法的操作条件、系统适用性和报告方式不会发生显著变化。当执行变更的风险对于类似多个产品相同或相似时,MAH应根据风险进行备案或申报,同类品种已获准的变更信息可作为平台分析方法的支持性数据,同时还应开展产品针对性的分析方法研究。
对于在多个国家获批上市的产品,往往面临着全球监管策略的差异和复杂性,MAH 在执行变更时需要充分了解各个国家/地区监管要求的差异,合理布局和规划在各个批准国家/地区的申报策略。
参考文献
[1] ICH Q6B, Specifcations :test prcedures and acceptance criteriafor biotechnological/biological products[S]. 1999.
[2] 李敏,常卫红,生物制品质量标准研究与建立一般原则的探讨[J],中国新药杂志,2017,26(16):1887-1893.
[3] PARR MK,SCHMIDT AH. Life cycle management of analyticalmethods[」l.JPharm Biomed Anal,2018,147:506 -517.
[4] VOLTA ESL,GONCALVES R,MENEZES JC,et al. Analyticalmethod lifecycle management in pharmaceutical industry: a re.view[Jl. AAPS Pharm Sci Tech,2021.22(3):128.
[5]李怡君,何伍,魏开坤.生物制品分析方法的生命周期管理[J].中国新药杂志,2023,32(2):181-188.
[6] ICH Q10.Pharmaceutical quality system[S].2008.
[7] ICH Q8( R2). Pharmaceutical development[S].2009
[8] ICH Q9( Rl). Quality risk management[S].2023.
[9] VERCH T, CAMPA C, CHÉRY CC,et al. Analytical quality bydesign, life cyele management, and method control[ J]. TheAAPS Joual,2022,24(1):34.
[10] ICH Q12. Techinical and regulatory considerations for pharma-ceutical prduct lifecycle managementS .2019.
[11] ICH Q14. Analytical procedure development ( draft version)[S].2022.
[12] THAKUR A,NAGPAL R,GHOSH AK,et al. Identification ,characterization and control of a sequence variant in monoclonaantibody drug product: a case study[J]. Sci Rep, 2021, 11(1):13233.
[13]陶磊,饶春明.重组蛋白药物制品相关蛋白的分析与评价[J].药物分析茶志,2018.38(11):1854-1864.
[ 14] 邓钦培,安然,徐通泽,等.毛细管电泳用于测定人绒促性素的解离亚基及其质量控制[J].药学学报,2017,52(3):430-435.
[15] 武刚,于传飞,王文波,等.成像毛细管等电聚焦电泳分析单克隆抗体电荷异质性的方法学验证及系统适用性对照品的制备[J].药物分析杂志,2019,39(1):62-69.
[16] LI M,ZHA0 X,SHEN D, et al. ldentification of a monoclonaantibody clipping variant by crss-validation using capillary elec.trphoresis-sodium dodecyl sulfate , capillary zone electrophoresismass spectrmetry and capillary isoelectric focusing-mass spec.tmmetry[J].JChromatogr A,2022,1684:463560.
[17] ZHANG X,CHEN T,LIV,et al. Cutting-edge mass spectrme.try strategy based on imaged capillary isoelectric focusing(iclEF)technology for characterizing charge hetemgeneity olmonoclonal antibody」. Anal Biochem,2023.660:114961.
[18] JONES M,PALACKAL N, WANG F,et al.“ High-risk” hostcell proteins(HCPs):A multi-company collaborative view[J]Biotechnol Bioeng,2021,118(8):2870-2885.
[19]ABOULAICH N, CHUNG WK, THOMPSON JH, et al. A novelapprach to monitor clearance of host cell prteins associated withmonoclonal antibodies[J]. Biotechnol Prog,2014,30(5)1114 -1124.
[20] BRACEWELL DG,FRANCIS R,SMALES CM. The future ofhost cell protein(HCP)identification during process develop.ment and manufacturing linked to a risk-based management foltheir control[Jl. Biotechnol Bioeng,2015,112(9):17271737.
[21] PILELY K,NIELSEN SB,DRABORG A,et al. A novel ap.proach to evaluate ELISA antibody coverage of host cell prteinscombining ELSA-based immunocapture and mass spectrometry[I].Biotechnol Prg,2020,36(4):e2983.
[22]WALDERA-LUPA DM, JASPER Y, KOHNE, P, et al. Host cellprotein deteetion gap risk mitigation: quantitative AC-MS fo)ELISA antibody reagent coverage determination[Jl. mAbs.2021,13(1):1955432.
[23]STEELMAN SL,POHLEY FM. Assay of the follicle stimulatinghormone based on the augmentation with human chorionic gona.dotropin[1l. Endocrinology,1953,53(6):604-616.
[24] NEVELLIF, PALMESE A, GLEIXNER R,et al. Biological assay to detemmine gonadotropin poteney: from in vivo to in vitrosustainable method」. Int JMol Sci.2023.24(9):8040
[25] 孙爽,王绿音,李品,等.基于报告基因的重组人促卵泡激素Fc融合蛋白生物学活性测定方法研究[J].药物分析杂志,2022,42(1):60-67.
[26]ENGLAND MR,STOCK F,GEBO J,et al. Comprehensive evaluation of compendial USP〈71〉,BacT/Alert dual-T, andbactec FX for detection of prduct sterility testing contaminants[1].JClin Microbiol,2019,57(2):e01548.
[27]KHUU HM,STOCK F,MCGANN M,et al. Comparison of auto-mated culture systems with a CFR/USP-compliant method for ste-rility testing of cell-herapy prducts[1]. Cytotherapy, 2004, 6(3):183 -195.
[28] PARVEEN S,KAUR S, DAVID SA, et al. Evaluation of growthbased rapid micmbiological methods for sterility testing of vac-cines and other biological products [J]. Vaccine,2011,29(45):8012-8023.
[29]李怡君,何伍,魏开坤,生物制品分析方法的生命周期管理[J].中国新药杂志,2023,32(2):181-188
[30] ERMER J,AGUIAR D,BODEN A,et al. Lifecycle manage-ment in pharmaceutical analysis: How to establish an effcientand relevant continued performance monitoring prgram [j]..Phar Biomed Anal, 2020,181: 113051.
[31] RUESCH MN,BENETTI L,BERKAY E,et al. Strategies forsetting patient-centrie commercial specifications for biotherapeuticproducts[Jl.JPharm Sci,2021,110(2):771 -784.

来源:中国新药杂志


