您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-07-02 08:40
生物类似药(biosimilar)主要指生物制品与原研药物在质量、有效性和安全性方面的表现是一致的。当然,如果安全性比参照药物更好,也是允许的。为了证明与原研药的一致性,需要开展CMC、非临床和临床的头对头对比研究。生物类似药评价遵循逐步递进(Stepwise)原则,从总体证据来判定相似程度,如下图所示,研究顺序从药学对比、非临床研究、临床药理研究和有效性/安全性研究。研究内容和程度取决于“上一步”对比研究的结果。

目前WHO、美国FDA、欧洲EMA、中国NMPA、俄罗斯、加拿大BGTD、巴西ANVISA、印度CDSCO、土耳其TMMDA、墨西哥COFEPRIS、南非SAHPRA等均发布了biosimilar指导原则。本文挑选主要法规国家biosimilar指南中对非临床部分的要求进行对比,并分享若干案例。
WHO、EMA、FDA及NMPA等均提到biosimilar产品非临床评价的策略是“逐步递进原则”。非临床评价的主要目的是解释药学比对中出现的差异。EMA、WHO、BGTD、CDSCO、TMMDA认为体内研究开展与否要视体外研究结果而定。如果结构、功能等药学比对相似,FDA认为仅需开展有限的毒理学研究即可。ANVISA则坚持在相关动物种属中开展体内非临床研究包括药效学研究、重复给药毒理学研究(含毒代动力学),并提交给监管机构。下面按照先体外、后体内的顺序做下详细介绍。
体外研究
EMA
体外研究的有些结果可能来自质量研究部分。总体上讲,体外试验至少应该包括:1)与靶点的结合;2)信号转导和细胞功能研究。关于“biological activity”and “immunochemical properties”,EMA将其归在“quality issue”中进行研究,但其实这部分也属于非临床体外研究的一部分,只不过在早期工艺研究和质量研究阶段,就需要对这两类属性进行表征,并证明与参照药物的一致性。
如果是抗体类生物制品,还需要开展Fcγ受体结合试验(FcγRⅠ、FcγRⅡ、FcγRⅢ)、FcRn及补体C1q结合。此外,围绕供试品的功能,也需要开展相关研究,如配体或受体阻断、激活。视抗体类型,对于膜靶点,建议开展Fc相关的功能(如ADCC、CDC)研究。组织交叉反应试验通常无需开展。
参比药物是具有批间差异的,且检测方法也有一定变异度,故建议采用多个批次进行对比研究。
FDA
FDA也是把“functional activities”and“receptor binding and immunochemical properties”放在质量部分进行描述。FDA思路与EMA类似,但特别强调如果供试品是多功能的,比如既有酶活又有受体介导的活性,应该对每项功能均进行对比研究。如果供试品是单功能,但有多个指标可评估其功能,比如酶动力学或与凝血因子的相互作用,则建议对每项指标均进行对比研究。FDA鼓励开发低变异度、高灵敏度的功能性实验方法,高变异度的实验方法价值很有限。
WHO、中国NMPA、南非SAHPRA、土耳其TMMDA的要求与EMA类似。加拿大BGTD仅提及建议开展非临床体外研究,但未详细描述推荐的具体试验。
巴西ANVISA、俄罗斯、NMPA、墨西哥COFEPRIS未明确体外研究的具体内容。印度CDSCO建议开展与参照药物对比的体外细胞功能试验如细胞增殖、配体/受体阻断。
体内研究
EMA
体内研究开展与否取决于体外对比研究的结果。如果拟开发产品在结构、质量、制剂(如新的辅料)等方面与参照药物有区别,将作为体内研究开展与否的关键证据。如果没有合适的体内研究动物模型,可以直接进入临床研究。如果理化特征、生物学特征以及非临床体外研究均显示与参照药物没有区别,则无需开展动物研究,可直接进入临床。
如果需要开展体内动物研究,则尽可能在每项研究中获得更多信息。
PK/PD:建议开展人体剂量下的量效关系研究,并与参照药物进行对比。
重复给药毒性研究:至少开展单剂量、单性别、无恢复期的重复给药毒理研究。如果只开展1个剂量,EMA建议选择剂量探索中的高剂量。EMA不建议在非相关动物中开展重复给药毒理研究。此外,原则上,EMA也不建议采用非人灵长类开展重复给药毒理研究。
免疫原性:虽然动物免疫原性不能用于预测人体情况,但可以用于对PK/TK的解释,建议采集,根据PK/TK的需要再决定是否检测。
安全药理学、生殖毒性和致癌性研究不需要开展。仅在出现新的辅料时,开展局部耐受性,且可以伴随在其它体内研究中开展。
WHO
PK和/或PD:建议开展临床拟用暴露量下的对比研究,包括定量的PK、PD。PD marker或有效性终点可以在疾病动物模型中考察。
重复给药毒性研究:不推荐在非人灵长类中开展。如果能合理说明,可以仅开展单剂量、单种属、无恢复期、仅in-life观察(无解剖及病理)的简化版毒理研究。应选择已知参照药物的高剂量进行对比。建议检测TK。除非因为杂质问题,想探索非特异性毒性,原则上不建议在非相关动物种属中开展重复给药毒性研究。
免疫原性研究:WHO建议采集ADA样本,用于对TK/PK的可能解释。
安全药理学、生殖毒性、遗传毒性和致癌性研究不需要开展。视新辅料的临床使用经验决定是否开展局部耐受性,且可以伴随在重复给药毒性试验中开展。
FDA
如果结构和功能学研究结果显示供试品的安全性尚有不确定性,FDA认为开展毒理学研究是有必要的。体内研究应按照ICH S6要求开展。毒理研究内容和程度取决于供试品和参照药物的体外对比研究的相似程度。FDA建议企业就是否开展动物试验及拟开展程度与监管机构进行沟通,且沟通节点应该尽早。
如果结构和功能试验结果显示与参照药物是相似的,则仅需要开展有限的动物试验即可,不需要处死动物,仅收集in-life指标、PD、PK及免疫原性。
反之,如果前期研究数据不充分,或前期结果提示产品质量有担忧,则需要开展标准的毒理研究,包括病理学、PD、PK、免疫原性等,具体参照ICH S6。
如果没有合适的相关种属,FDA认为在非相关种属中开展毒理研究是没有意义的。但有些试验还是可以开展的,比如与参照药物比较的PK。另外,对于无法开展动物毒理研究的情况,FDA建议采用人的细胞或组织开展体外对比研究。
如果结构和功能试验、动物毒理试验结果显示与参照药物相似,则不需要开展安全药理学、生殖毒性和致癌性试验。
FDA鼓励至少在1个动物种属中伴随PK、PD指标考察,以增加供试品与参照药物相似的总体证据。
关于免疫原性研究,FDA认为虽然动物ADA结果不能反映人体,但如果供试品与参照药物生产过程存在差异,比如杂质或辅料,则可能体现在ADA的不同。此外,供试品和参照药物如果结构或功能存在差异,也可能出现ADA的不同,而这些是其它分析方法所不能代替的。
FDA对动物研究数据的要求示例如下。

中国NMPA
对于药学比对无差异或很小差异的,可以简化体内研究,仅开展药效、药代和免疫原性对比研究。反之,则需要进一步开展体内药效和毒理比对研究。体内药效比对试验研究应尽可能选择参照药采用的相关动物种属和模型。药代动力学应在相关动物种属中开展,单次和多次药代均需要进行,且单次给药(多个剂量组)PK需要单独开展。多次给药PK可以伴随药效学或毒理学试验开展。免疫原性研究也是NMPA关注的重点指标,可结合在多次给药PK或毒性试验中开展。重复给药毒性试验开展与否视药学、PD、PK、免疫原性等结果而定。如果开展,通常进行一项相关动物种属的至少4周的研究,伴随TK、局部耐受性等。
加拿大BGTD
该机构认为如果前期研究已经证明了与参照药物的相似性,可以不开展体内研究。对于需要开展体内研究的情况,未披露更进一步的细节要求。不过,对于安全药理学研究、生殖毒性、遗传毒性和致癌性研究不作要求,可以不开展。
其他国家
巴西ANVISA强制要求开展目标适应症的PD研究和重复给药毒性研究并伴随毒代动力学检测。俄罗斯和墨西哥就没有这类要求。
南非SAHPRA要求开展体内动物试验,包括至少1个重复给药毒理研究和免疫原性研究。其它毒理试验通常不需要开展。
印度CDSCO认为前期研究如果证明与参照药物相似,则不需要开展体内药效学研究。反之,则需要开展。如果开展重复给药毒理研究,建议至少采用临床等效剂量,选择相关动物种属,给药周期不少于28天,14天的恢复期。CDSCO对重复给药试验组别的要求如下图所示。

CDSCO建议开展免疫原性对比研究,可以伴随重复给药毒性试验开展。其它毒理研究如安全药理学、生殖毒性、遗传毒性、致癌性、局部耐受性要求同WHO。
土耳其TMMDA要求与EMA一致。
案例
生物类似药的非临床研究案例注定没有统一模板,因为非临床研究的内容多少和程度完全取决于自制品与参照药物之间的药学、生物学等体外对比研究结果。二者区别越小,对非临床研究的需求也就越小,试验内容也就越少。反之,则需要开展的非临床内容会多上不少。目前已经上市的生物类似药很多,选择几个代表性案例,略作分享。
Amjevita
Amjevita又名ABP501,是由Amgen开发,仿制的前药王阿达木单抗(商品名:Humira),是一款TNFα抗体,于2015年11月25日在美国FDA递交BLA申请。
ABP501体外药理开展了与US-Humira详细对比的生物学和免疫化学表征包括与可溶性TNFα结合试验,凋亡抑制试验,FcγRⅢa 158V、FcγRⅢa158F、FcγRⅠ或Ⅱa结合试验,ADCC、CDC、C1q结合、FcRn结合、与膜表面TNFα结合试验、抑制TNFα诱导的IL-8试验、抑制TNFα诱导的趋化作用等。所有这些功能性试验均达到了预设的相似性标准。设计的供试品和原研品批次,少则3批,多则20多批。关键性的试验,比如靶点结合、靶点功能抑制、Fc功能等采用的批次≥10批。当然,这是递交BLA的数据,IND阶段一般还没有生产那么多批次。
ABP501非临床仅开展了两项研究,一项是ABP501与US上市的Humira食蟹猴TK对比研究,一项是食蟹猴毒理对比研究(伴随TK)。有些人可能比较好奇,第2个试验不是可以覆盖第1个吗?真实情况是,本来确实计划仅开展一项动物试验,结果第1个试验剂量设计失误,剂量太低,选用的原研品Humira重复给药毒性试验的低剂量(无论EMA还是WHO均建议选择高剂量),给了2次药就停了,收集了in-life指标,主要用于TK对比。之后,重新开了第2个毒理试验。第2个试验的设计如下,评价指标包括死亡率、临床观察、体重、摄食量、眼科、ECG、临床病理、大体解剖及组织病理学、TK和ADA检测。从下表不难看出,该试验是一项简化版的毒理研究,属于单剂量、单种属、双性别(第1个试验是单性别)、无恢复期的设计。当然,WHO和FDA对于简化版毒理方案,还可以允许动物不处死,仅进行in-life指标收集。不过,ABP501是做了完整的组织病理学检查的。
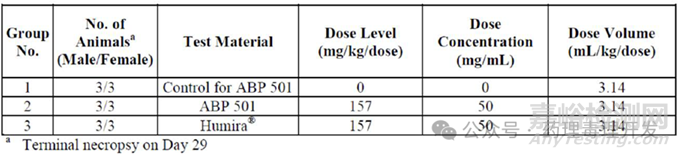
ABP501未开展体内药效学、单独的药代动力学、遗传毒性、生殖毒性和致癌性相关的研究。
Mvasi
Mvasi是由Amgen开发的VEGF抗体Bevacizumab的biosimilar。VEGF主要结合血管内皮细胞表面的VEGFR1和VEGFR2,诱导细胞增殖和血管生成。
质量部分与体外药理有关的研究包括:1)VEGF增殖抑制试验;2)VEGF165/121结合研究(ELISA、SPR);3)VEGFR2激酶磷酸化抑制研究;4)VEGFA、C、D结合特异性研究;5)FcγRⅢa 158V、FcγRⅢa158F、FcγRⅠ、FcγRⅡa、FcγRⅡb结合试验, C1q结合、FcRn结合、ADCC、CDC。FcγRⅢa 158V结合结果未符合预设相似性标准,但考虑到本品无ADCC活性,所以这一差异不具备临床意义。其它体外研究结果均符合预设的相似性标准。
为进一步证明与Bevacizumab的相似性,Mvasi非临床研究开展内容如下:
药效学研究:1)A431异源移植瘤模型中的体内药效对比(各1个剂量);2)Colo205异源移植瘤模型中的体内药效对比(各1个剂量);3)抑制VEGF诱导的雌性无胸腺小鼠血管渗漏试验(各4个剂量)。
药代动力学研究:雄性SD大鼠单次、单剂量、IV给药PK对比研究(1mg/kg、12只/组)。
毒理学研究:1个月食蟹猴毒理对比研究。供试品和参照品各1个剂量(50mg/kg),6只/组,雌雄各半,每周给药2次,给药4周。本研究也属于简化版研究,单剂量、单种属、双性别、无恢复期的设计,与ABP501设计类似,毕竟同一家的产品。
Trazimera
Trazimera是HER2抗体trastuzumab的biosimilar,由辉瑞公司开发,2018年7月在欧盟获得批准上市,2019年3月获FDA批准上市。
Trazimera开展了与EU和US两个产地trastuzumab的体外功能和结合试验。在HER2靶点结合、HER2下游信号通路阻断方面,三者表现相似。ADCC(NK作为效应细胞)、ADCC(PBMC作为效应细胞),FcγRⅢa 158V、FcγRⅢa158F、FcγRⅡa 131H、FcγRⅡa 131R、FcγRI、FcγRIIb、FcγRIIIb、FcRn、C1q结合也是相似的。
Trazimera开展的非临床研究很少。体内药效学研究、单次给药毒性试验、遗传毒性研究、生殖毒性研究和致癌性试验均未开展。
开展了Trazimera、trastuzumab-US、trastuzumab-EU,各3个剂量,小鼠单次给药TK研究,伴随抗药抗体检测。这个试验也不是辉瑞主要开展的,是IND时监管机构要求其补充的试验。虽然trastuzumab不识别小鼠neu受体(HER2同源受体),小鼠不是相关种属,但可以比较三个药物的清除和分布。
Trazimera还开展了小鼠2周重复给药毒理研究。这个试验的目的主要是考察杂质可能引起的毒性。未设置原研品对照。当然,小鼠不是相关种属,在这个体系中与原研品对比毒性也没有意义。
所以,实际上Trazimera只开展了一项啮齿类动物的单次给药PK对比研究。
CD20单抗
这个案例以CDE发表的《从抗CD20单抗探讨我国生物类似药非临床研究与评价的思路》一文为主要参考,看下我国药品监管机构对生物类似药的审评尺度和大致思路。
抗CD20 单克隆抗体是一类治疗B 细胞淋巴瘤的靶向药物,原研药为1997 年罗氏制药有限公司上市的利妥昔单抗(rituximab,商品名为美罗华)。利妥昔单抗与B 细胞上的CD20 抗原结合后,通过ADCC或CDC介导B 细胞溶解的免疫反应。国内申报利妥昔单抗生物类似药的案例很多,CDE举例如下:
案例A
申请非霍奇金淋巴瘤适应症。体外药效开展了与细胞表面CD20亲和力试验、ADCC、CDC、组织交叉反应对比研究。体内药效开展了猴单次和多次PK/PD研究,以B细胞清除作为PD指标,并与参照药物进行对比。体内、体外结果均显示与参照药物相似。
伴随体内药效开展了单次、多次PK研究,并与单剂量利妥昔进行对比。另外,伴随重复给药毒性试验开展了多次给药的TK对比研究。证明了与参照药物的药代动力学相似性。
伴随重复给药毒性试验开展了比较详细的免疫原性和免疫毒性研究,包括IgG抗体水平、淋巴细胞数目等药效相关指标,以及抗体产生时间、抗体滴度、中和抗体等指标。结果显示与利妥昔单抗表现一致。
CDE认为案例A总体证据证明了与利妥昔单抗的相似性。
案例B
体内、体外药效开展的试验内容与案例A类似。但是,该企业除了肿瘤适应症,同时申请了类风湿关节炎(RA)适应症。而且,未提供RA中的体内药效对比数据。另外,该企业开展的重复给药毒性试验未与参照药物比对。所以,被CDE建议补充RA药效学、与参照药物对比的重复给药毒性试验和伴随的TK、ADA研究。
案例C
仅进行了单次给药PK研究。
进行了给药12周,恢复期8周的重复给药毒性试验。恢复期结束时,淋巴组织B细胞不能完全恢复至给药前基线值。恢复期设置过短。
未进行与利妥昔的免疫原性比对。无法判断二者免疫原性的差异。
CDE:未进行比对的重复给药毒性试验和免疫原性试验,相似性依据不足,建议进一步提供与RBP 比对的重复给药毒性试验、免疫原性以及多次给药的药动学试验。
案例D
进行了单次和多次给药PK研究,但未设置利妥昔对照。故,被CDE要求补充药动学单次、多次给药对比研究。
开展的猴重复给药毒理研究,但高剂量仅为1倍的临床拟用剂量。且动物体内检出ADA,进一步影响药物暴露。建议提高剂量或调整给药方式重新开展重复给药毒性试验。
CDE:本品质量研究未提供足够的一致性证据。如果按照生物类似药思路开发,应进行比对的非临床研究,以进一步评价与参照药的一致性,内容包括必要的比对的药效、药代和一种相关动物种属的重复给药毒性试验。
CDE这篇文章比较早,发表于2015年,企业开展试验的时间更早,那个节点,国内生物类似药刚刚扎堆兴起(以CD20、HER2、EGFR、TNFα、VEGF靶点为主),尚无成熟的国内外经验可以参考,非临床研究策略比较多样,很多企业还不太懂得如何开展生物类似药的非临床研究。随着各个国家监管机构指南的出台以及获批上市的生物类药物案例的增多,整个生物类似药的评价思路已经非常成熟,CDE的审评理念应该也在变化。所以需要注意以上案例的时效性。
最后
已经上市的生物类似药案例很多,篇幅所限不再一一举例。从法规角度看,体外研究部分,各国的要求其实差不多,结合拟开发产品的MOA、靶点结合、信号通路、功能等开展一系列敏感、特异的多批次自制品和原研品的对比研究。区别出现在体内研究部分。对于前期药学和体外研究结果依然存在担忧或不确定性的情况倒还好,开展比较全面的药效、PK和毒理对比。对于前期研究证明比较相似的情况,EMA、FDA允许开展简化的毒理设计,比如单种属、单剂量对比、无恢复期等。Amgen的Amjevita和Mvasi就属于这类设计。辉瑞的Trazimera仅开展了单次给药、三剂量的小鼠PK对比研究,未开展毒理对比。如果在国内或者印度等国家申报,采用简化方案设计时,建议先与监管机构进行沟通确认。

来源:药理毒理开发


