您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-07-08 08:20
摘 要 Abstract
病原宏基因组高通量测序(mNGS)技术因其检测时间短、分辨率高,能识别罕见、新发病原体引发的感染或者混合感染等优势,已经被广泛地应用于临床疑难感染的辅助诊断。然而,由于目前尚缺乏标准化生物信息学分析流程和高质量临床微生物基因组参考数据库等问题,一定程度上制约了该技术临床应用的进一步发展。本文介绍了高质量临床微生物基因组参考数据库建设现状,探讨了高质量临床微生物基因组参考数据库构建的技术要求、质量控制过程和实现方式,并提出相关思考和建议。
Pathogen metagenome next-generation sequencing (mNGS) technology has been widely used in the detection of clinical infectious diseases due to its advantages of short detection time, high resolution, and the ability to identify infections caused by rare and emerging pathogens or mixed infections. However, the lack of standardized bioinformatics analyses and high-quality clinical microbial genome reference databases has restricted the further clinical application of this technology. This paper introduces the current status of high-quality clinical microbial genome reference database construction, discusses the technical requirements, quality control processes, and implementation methods building such a database, and provides related
thoughts and suggestions.
关键词 Key words
病原宏基因组高通量测序;生物信息学分析;微生物基因组参考数据库;建设现状;技术要求
metagenomic next-generation sequencing; bioinformatics analyses; microbial genome reference database;construction status; technical requirements
感染性疾病对人类健康构成重大威胁,其病原体呈现多样化和复杂化的发展趋势。快速、准确地进行病原检测,对于临床诊断具有重要意义。病原宏基因组高通量测序(metagenome next-generation sequencing,mNGS)技术已经从科研领域走向并广泛地应用于各种疑难感染病原微生物检测、新发突发传染病病因分析和溯源、毒力耐药基因检测等领域。常规的病原微生物培养法、血清学方法、抗原/抗体检测以及传统核酸检测技术,由于耗时长、灵敏度和特异性低、检测谱窄等原因,在上述应用场景下受限明显[1]。
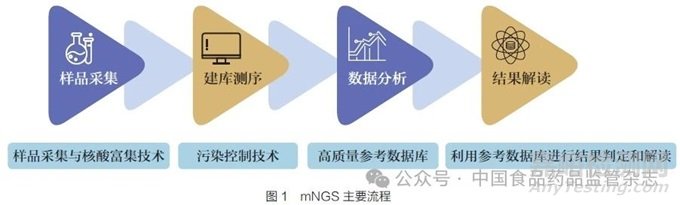
mNGS 技术可以直接对临床样本中的所有核酸进行无偏倚测序和分析[2],包括样品采集、建库测序、数据分析、结果解读(图1)。与传统的基于培养或分子的方法相比,mNGS 技术无需进行病原分离培养或设计引物及探针,应用高通量测序平台,可以在10~30h 完成测序。通过分析测序数据,不仅能够对样本中的细菌、真菌、病毒和寄生虫等物种和丰度进行检测,同时还能够分析其携带的耐药和毒力基因[3]。
尽管mNGS 技术已经在临床感染诊断领域得到了广泛应用,但仍存在较多技术挑战[4-6]。在实验过程中,由于测序和分析的是样本中的全部核酸,其中必然会携带来源于宿主、试剂、耗材、实验室环境和人员等核酸,可能对生物信息学分析造成干扰。一方面,需要建立标准化的实验操作流程并严格执行,以保证测序数据的质量;另一方面,需要设置科学合理的阴阳性判断阈值,建立标准化的生物信息学分析流程,以及高质量的微生物基因组参考数据库对测序数据进行处理和分析,以实现准确鉴定病原的目的[7]。
本文介绍了高质量临床微生物基因组参考数据库建设现状,并从技术要求、质量控制过程和实现方式等方面,综述了构建高质量临床微生物基因组参考数据库面临的挑战,并提出了相应建议。
1、 高质量临床微生物基因组参考数据库建设现状
mNGS 技术是无预设、无偏倚的测序,结果的分析和报告依赖于测序序列与基因组参考数据库的比对,因此mNGS 检测性能在很大程度上取决于生物信息学分析的分类算法组合,以及基因组参考数据库。该数据库主要由两部分组成,包括宿主基因组数据库和微生物基因组参考数据库,其中后者对报告结果准确性的影响更直接且关键。中华医学会检验医学分会发布的《宏基因组测序病原微生物检测生物信息学分析规范化管理专家共识》[8] 中,建议满足临床检测的比对数据库,至少应包括微生物、人源及背景序列数据等;同时建议实验室应保证数据库稳定运行,及时补充新发病原体,提高数据库覆盖度。
然而,关于如何构建适用于mNGS 技术的高质量临床微生物基因组参考数据库,尚缺少针对性的技术文件和共识。目前,国内外没有可以直接适用于mNGS的微生物基因组参考数据库,科研机构、临床实验室以及mNGS产品研发机构等,需要利用公共数据库中的基因组数据,自行构建用于比对的数据库。不同机构自建的参考数据库,所选取的微生物基因组数据的来源、质量控制标准、清洗流程及质量均不一致, 可能会出现不同机构应用mNGS 检测的结果参差不齐、可比性较差的情况。微生物基因组参考数据库的建设是一个复杂的过程, 因此, 应尽量形成一套公认的、详细的指导方案, 并且在建设的过程中要考虑尽量保证准确、高效且对临床具有可操作性。
2、 高质量临床微生物基因组参考数据库建设技术要求
建设用于mNGS 结果比对的临床微生物基因组参考数据库,可以从数据源、代表性基因组的选取,以及数据质量控制等方面来进行规范(图2)。

2.1 数据源选择
国际核酸序列数据库联盟(International Nucleotide Sequence Database Collaboration,INSDC),是由美国国家生物技术信息中心(National Centerfor Biotechnology Information,NCBI) 建设的基因数据库(GenBank)[9]、欧洲核酸库(European Nucleotide Archive,ENA)[10] 和日本DNA 数据库(DNA Data Bank of Japan,DDBJ)[11] 组成,收录了绝大多数全球科学家发表的基因组原始测序数据、基因组拼接数据以及蛋白质序列数据等组学数据。三大核酸数据库定期进行数据交换,以保证数据存储标准和内容的一致性。其中GenBank 数据库是选取参考基因组的主要数据来源。
由于INSDC 数据库中收录的数据主要来源于全球用户的直接提交,因此存在大量序列数据质量低、物种注释不准确及序列污染等质量问题。因此,为了提供高质量的参考数据,除了GenBank,NCBI 还建立了Taxonomy[12]、RefSeq[13]、Pathogen Detection[14] 等数据库。Taxonomy 数据库是关于物种分类和命名的数据库,目前收录超过7万个物种的名字和种系,并且每一条记录都链接到其对应的核酸或蛋白序列。需要注意的是,NCBI 维护的分类学数据库是基于序列的进化关系建立的分类地位,与一些分类学及命名数据库如原核生物名称列表(List of Prokaryotic nameswith Standing in Nomenclature,LPSN)数据库在物种分类和命名上有部分冲突。因此,在确定基因组对应的物种正确的分类和命名时,还需要参考标准的分类学数据库。RefSeq 数据库是经过NCBI筛选过的非冗余数据库,具有较高的准确性。因此,如果某个物种具有RefSeq 基因组时,可以优先选取该基因组序列。Pathogen Detection 是病原微生物专题数据库,整合来源于食品、环境和患者的细菌病原基因组数据,同时还对集成的数据提供聚类分析,从而用于识别潜在传播链,并且利用AMRFinderPlus[15] 工具通过使用蛋白质注释或组装核苷酸序列来识别耐药基因和相关突变位点。
除NCBI 以外,国际微生物领域中一些研究机构或组织,为了支撑高质量的数据研究进行了大量的基因组测序,并同公共来源的数据整合形成了专题参考数据库, 例如微生物基因组和微生物组综合数据库(Integrated Microbial Genomes & Microbiomes,IMG/M)[16]、美国食品药品监督管理局参考级微生物测序数据库(Food and Drug Administration-Database for Reference Grade Microbial Sequences,FDAARGOS)[17]、细菌和病毒生物信息学资源中心(Bacterial and Viral Bioinformatics Resource Center,BV-BRC)[18]、全球模式菌株目录(Global Catalogueof Type Strain,gcType)[19]等。IMG/M 由美国能源部联合基因组研究所建设,整合了微生物基因组、宏基因组和病毒组数据,并提供在线的分析平台。FDA-ARGOS 数据库是一个经过注释的高质量测序病原微生物基因组数据库。BV-BRC 由美国国家过敏和传染病研究所建立,整合了细菌、流感研究数据库和病毒病原体数据库与分析资源的资源,以帮助研究人员分析不断增长的基因组序列和其他组学相关数据。gcType 是由我国中国科学院微生物研究所国家微生物科学数据中心(National Microbiology Data Center,NMDC) 建立和维护,对全球所有细菌和古菌的模式菌株进行基因组测序,目前该数据库已经整合了15 823 个种的原核微生物模式基因组序列,其中自测基因组4805 个,极大地填补了公共数据源中的空白。
当机构自建参考数据库,在选择数据源时,要对不同来源的相同数据进行去除冗余的处理,或者明确不同数据库中,各类数据的来源,避免重复选择以减轻工作量。为此,NMDC 建立了一个开放的基于人工注释的全球病原体目录(Global Catalogue of Pathogens, gcPathogen)[20],旨在支持快速和准确的病原体基因组分析、流行病学研究,以及抗生素耐药性特征和毒力因子的监测。病原体清单来自医学或政府病原体清单和出版物的证据支持的数据,包括来自509 种细菌的110 万个基因组、来自407种真菌的6785 个基因组(其中30% 以上来自自测真菌基因组数据)、来自226 种病毒的9 万个基因组(不包括流感病毒、新冠病毒基因组数据)、来自174 种寄生虫的670 个基因组数据。
尽管从国际公共来源的数据库可以实现绝大多数微生物基因组数据的整合,但目前还有大量的未测序物种,一个尤为突出的情况是目前国际数据库中,感染性真菌基因组数量较为稀少。根据统计,有明确感染史的致人类感染的真菌超过400 种,但目前公共数据库中,有基因组测序数据的仅有不到300 种。还有许多重要的物种,例如在《WHO 指导研究、开发和公共卫生行动的真菌优先病原体清单》[21] 中列为第二级重要性的毛霉属,大部分具有感染史的物种没有基因组数据。这些问题对于相关病原体的检出造成巨大挑战。因此,在整合公共数据的同时,进行病原微生物基因组测序,补充常见但目前缺失的病原体基因组数据,也是实现临床微生物基因组参考数据库完整性的一个重要方面。
2.2 基因组数据质量控制
公共来源的微生物基因组数据来自全球各地用户的提交,可能存在序列污染、物种分类错误、命名不准确、测序数据质量低、完整度差等各种质量问题。因此,对数据进行质量控制处理与整合,是建设高质量的微生物基因组参考数据库最重要的步骤,建议考虑且不限于以下数据质量控制方法。
(1)分类信息评估与确认。存在于公共数据库中的数据,可能由于提交人的失误,或受限于提交时可进行比对的参考数据较少,存在分类错误。因此,对于通过质量控制的序列, 应当进行平均核苷酸相似度(average nucleotide identity,ANI)比较,或者构建进化树,确定每一个挑选的基因组具有正确的分类地位,剔除分类错误基因组。
(2)测序质量、组装质量评估。测序深度不足、组装不准确等因素造成的基因组序列不完整,或基因组序列片段化严重(N50小,contigs 或scaffolds 数量过多)等。利用CheckM[22] 等软件对数据进行质量评估,在有多条序列可选择的情况下,优先选择测序质量好、完整度高的序列,比如污染度小于1%,完整度大于99% 的基因组数据。
(3)污染序列过滤。在微生物全基因组测序的过程中,微生物样本、核酸提取建库过程都有可能受到人类基因序列的污染。在mNGS 分析中,如果人源序列过滤不干净,残留的人源序列就可能被错误地鉴定到了这些含有人源基因片段污染的微生物,造成假阳性检出。因此,需要去除宿主序列等污染再进行数据库的构建,减少比对假阳性的结果。
(4)微生物命名准确性。微生物命名需要参考国际权威的微生物命名数据库, 细菌可以参考LPSN[23]、真菌可以参考真菌命名(Fungal Names)[24]、病毒可以参考国际病毒分类委员会(International Committee on Taxonomy of Viruses, ICTV)[25]等数据库。然而,无论哪类数据库,微生物的命名都会随时更新曾经分类错误的数据,包括分类地位的改变或者命名的改变。因此,保持参考数据库的定期更新,是保证数据库质量的一个重要因素。
2.3 代表性基因组选择
高质量的参考数据库既要做到全面,能够提供不同分类等级的微生物基因组的代表性特征;又要做到简洁,太多的冗余序列会造成分析时间增加,或可能导致假阳性和假阴性问题而影响临床使用效果。
在选择代表性基因组时,需要充分考虑鉴定的目的和不同微生物的特点。如果缺失了病原微生物的基因组数据,可能导致测序序列无法比对,造成假阴性结果;如果缺失了非病原微生物的基因组数据,则可能导致测序序列错误比对到该缺失基因组近缘的病原微生物,而造成假阳性结果。对于病原微生物,为了提供准确和高分辨率的鉴定结果,可选择不同血清型、基因型的代表性基因组。对于病毒数据,由于序列较短,且病毒分型较为复杂,在选取参考基因组时,应尽量全面地纳入代表性数据,甚至应包括部分不完整但能提供分型特征基因序列,从而实现更加准确的鉴定。
此外,有一些物种的种内基因组差异较大,或者与其他近缘物种在基因组特征上重叠情况严重,如金黄色葡萄球菌与凝固酶阴性葡萄球菌、大肠埃希菌与志贺菌、鼻病毒的不同株系等。对于这些物种,选取代表性基因组无法代表种内所有的基因特征,或无法实现与近缘种的区分。这类问题的解决方案,可以考虑通过构建进化树或基于基因组相似性进行聚类分析,选取不同进化分支或聚类的代表性基因组;或者可以构建种内共性及特异性基因的泛基因组集,通过比对的方式来代替单个代表性基因组的选择。
3、 高质量临床微生物基因组参考数据库的相关数据库建设
3.1 人源参考基因组数据库
常用的人基因组包括Hg19、GRCH38 和YH2.0, 以及由国际科学团队端粒到端粒联盟(Telomere-to-Telomere,T2T)于2022 年发布的完整无间隙的人基因组T2T-CHM13。建议实验室根据最新版国际人类参考基因组,构建全面特异的人源基因序列数据库。
3.2 微生物知识库
mNGS 的检测报告, 一般包括比对到病原微生物的种类、病原微生物是否超过阳性阈值条件等关键信息;还可以提供物种的相对丰度、重要的耐药和毒力基因,以及检出的物种中包含的病原微生物感染引起的临床症状等附加信息。因此, 机构自建mNGS 参考数据库时,在保证宿主基因组数据库和微生物基因组参考数据库质量的前提下,可以附加建立微生物知识库,从而为临床诊断与用药提供更丰富的参考信息。
例如,病原微生物知识库提供不同感染部位主要的定植菌、条件致病菌和感染菌的清单,同时提供该病原微生物或症候群感染相关的描述信息;耐药基因数据库可以整合来源于抗生素耐药综合数据库(Comprehensive Antibiotic Resistance Database,CARD)[26]、识别测序数据中耐药基因和表型预测资源库(ResFinder)[27] 和毒力因子数据库(Virulence Factor Database,VFDB)[28] 等国际公认数据库的数据。由于mNGS测序数据中微生物基因组序列占比相对可能较小,可能无法做基因组拼接,基于短测序读序数据无法判断所检出的耐药基因或毒力因子位于某一个具体的病原物种中。因此,可以建立耐药和毒力基因分布频率数据库,通过比对所关注的基因存在的频率,给出间接的参考信息。
3.3 背景微生物基因组数据库
mNGS 检测过程中, 由于采样和试验操作引入的实验室环境和操作人员携带的微生物、试剂原料中的潜在工程菌,以及在试剂和耗材生产过程中引入的生产环境和生产人员携带的微生物等,一般统称背景微生物或背景菌,可能对生物信息学分析环节造成影响[29]。不同样本中背景菌的组成、来源,以及背景菌对检测结果的影响等均复杂多变。构建背景微生物基因组数据库时,可以根据使用mNGS 的实验室具体操作流程、检测样本的具体类型和处理要求,结合前期研究的数据进行建设并监测;或者可以选择相应模式菌株的基因组或者RefSeq 数据库中的参考基因组。
4、 其他要求
临床微生物基因组数据库建立后的一个重要环节,是质量控制与评价。基因组数据库和生物信息学分析一起构成了mNGS 检测的非试验操作部分,即干实验环节。应用可溯源的数字参考品,可以对mNGS 干实验环节整体进行评价,评价内容包括物种鉴定的准确性、近缘微生物同源干扰影响和微生物种类完整性等;数字参考品结合高质量的基因组数据库,还可以对软件算法及其运行参数进行评价。由于各机构自建的参考数据库,都是经过与配套软件算法一同构建和优化的,难以对数据库进行单独评价。此外,目前尚无公认权威的标准化或国家级数字参考品,能够对参考数据库和生物信息学分析流程进行质量评价。
参考数据库的建设还需要考虑安全的要求,一方面,对于使用的临床样本数据,需要具备完整的伦理审查过程,保障自测基因组数据的安全性和可用性。另一方面,利用基于隐私计算和区块链结合的技术,通过单向隐私计算及多方安全计算等方式,在保障数据库安全的同时,为用户提供符合数据分类分级要求的使用平台。
通常公共数据的更新时间较为频繁,为保证数据准确性,对于基因组数据库和物种分类数据库应当对更新的数据进行跟踪及验证,以确保更新的内容对检测结果无影响,并根据评价的结果对数据库进行必要的更新。对于微生物知识库,应能够提供来源于公共数据库数据的更新时间和版本号。
5、 结语
当前,mNGS 技术广泛应用于临床感染诊断领域,但仍面临生物信息学分析流程和临床微生物基因组参考数据库标准化程度低的挑战。一方面,应当尽快建立自主知识产权的高质量临床微生物基因组参考数据库,并建立标准化的数字参考品和验证流程,实现对其质量的验证与评价。另一方面,由于微生物基因组参考数据库是影响软件性能的重要因素,因此,需要对数据库的全面性、准确性和代表性进行科学地测试与评价未来,可以探索使用数字参考品开展对数据库性能评测的研究。
数据安全和隐私保护越来越成为一个制约数据使用的重要因素,利用区块链、联邦学习、隐私计算等数据安全保护技术在保障数据安全的同时,提升数据的可用性,实现数据价值的挖掘,为临床诊断提供更多的参考,也是在当前国家大力发展数据战略的形势下,需要面临的思考和挑战。

来源:中国食品药品监管杂志


