您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-07-25 09:36
有关物质是药品的关键质量属性之一,HPLC是有关物质检查的主要方法, HPLC测定杂质含量最理想方法为杂质外标法,但杂质对照品往往不易获得,加校正因子的主成分自身对照法或主成分外标法应运而生,已成为各国药典中HPLC测定杂质含量的主要方法。
校正因子易受到多种因素影响,即使采用相同的分析方法,校正因子也可能因仪器、色谱柱、分离度等影响而存在差异,因此虽然法定方法收载了校正因子,亦需进行校正因子的验证或确认。当实测校正因子与药典存在较大差异时,应进行分析解释。
一、校正因子的定义
HPLC定量测定中的校正因子是某物质与所选定的参照物质的绝对校正因子之比,各国药典表述略有不同。当校正因子在0.2~5.0范围以外时,应改变检测波长或参照物并重新确立校正因子,或采用杂质外标法进行定量。
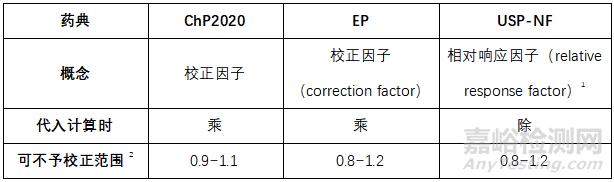
1相对响应因子与校正因子互为倒数关系。2即按校正因子1.0计;USP-NF 2022起,在通则<621> Chromatography中删除了当相对响应因子超出0.8~1.2范围时使用校正因子的表述。
二、校正因子的确立方法
HPLC校正因子的确立方法主要包括单点法、多点法和标准曲线法,其中标准曲线法是确定校正因子最常用的方法。
(1)单点法
制备适当浓度的杂质对照品溶液(cA)和主成分对照品溶液(cB),进样分析得到杂质峰面积(AA)和主成分峰面积(AB),按下式计算待测杂质的校正因子。
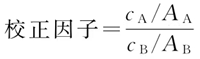
(2)多点法
制备至少涵盖定量限至杂质限度的多个浓度杂质对照品溶液和主成分对照品溶液,进样分析,按照单点法公式计算得到多个校正因子,取平均值即得。
(3)标准曲线法
制备至少涵盖定量限至杂质限度的系列浓度杂质对照品溶液和主成分对照品溶液,进样分析分别得到杂质和主成分标准曲线,主成分标准曲线斜率与杂质标准曲线斜率比值即为校正因子。
三、实测结果与药典不同的分析解释
校正因子研究是CDE发补的重点内容之一,近年来CDE尤为关注申报企业实测校正因子与药典收载值的一致性,例如以下发补意见:本品采用USP收载的有关物质分析方法,方法学验证结果显示杂质A和杂质F自测校正因子与USP差异大,请分析原因,并选择合适的杂质定量方法。
实测校正因子与药典收载值差异大的可能原因主要包括以下几点:
3.1 对照品赋值不准确
杂质对照品赋值准确与否直接影响校正因子值,由于杂质对照品纯化过程通常无法完全除去有机溶剂、水分、无机盐等杂质,因此应采用含量值(Assay)而非纯度值(Purity)进行校正因子计算。如对照品含量过低,应评估其适用性。
杂质对照品的定量方法主要包括质量平衡法和定量核磁共振法。由于杂质对照品不易获得,一般可采用TGA等微量方法替代传统的质量平衡法,再结合HPLC色谱纯度计算其含量。定量核磁共振法则利用1H-NMR谱中响应的积分值与化合物的质子当量成反比且与浓度成正比的原理,通过在样品中加入已知含量的内标物质,即可计算出所测定对照品的含量。1H-NMR法需要选择合适的内标物和参与定量的质子,以避开对照品中类似结构的杂质影响赋值准确性。
对照品赋值偏高将使得校正因子值偏大,反之则偏小。通常,优选法定来源且赋值明确的杂质对照品。需要注意的是,中检院、EDQM及USP部分杂质说明书中注明仅用于系统适用性或定位,应评估其含量的准确性。例如,某品种采用中检院杂质对照品进行研究,结果显示其校正因子为1.3,而USP收载值为1.1,CDE发补要求解释差异。企业采用1H-NMR定量法测定中检院对照品的含量为86.4%,而说明书中的含量值为98.9%,两者存在明显差异。按照核磁定量值计算,该杂质的校正因子为1.1,与USP一致。
3.2 对照品发生降解
对照品降解是造成实测校正因子与药典值不一致的重要原因。杂质对照品或杂质对照品所配制的对照品溶液发生降解,将使得校正因子偏大。主成分对照品或主成分对照品溶液发生降解,则将使校正因子偏小。
因此,对照品的保存应严格按照其贮藏条件,并关注对照品在贮藏过程的稳定性情况。部分对照品本身稳定性极差,此类对照品关注COA报告中的赋值时间,尽量采购近期新标定的对照品,并在采购后尽快完成研究。引湿性强的对照品吸潮后含量不再准确,且可能进一步降解,建议按小包装规格采购,开封后尽快使用并妥善保存,在称量过程应严格控制环境湿度。
酯类、内酯、酰胺等结构的物质在含水体系中的稳定性可能较差,应考察对照品在有关物质测定稀释剂中的稳定性情况,必要时更换适合的稀释剂,确保在考察期间溶液稳定,结果准确可靠。需要关注的是,不同浓度下的溶液其降解情况可能不一致,必要时应加以验证。
另一方面,由于杂质对照品稳定性问题或可及性问题,药典质量标准可能未进行校正因子验证,按默认校正因子1.0计。例如某品种多个杂质实测校正因子与USP差异较大,经了解,该品种USP质量标准由印度某企业参与起草,经联系该企业,其反馈其中3个特定杂质由于稳定性问题未进行验证,1个杂质由于未获得对照品而未进行验证,均按照默认值1.0定入USP。
3.3 杂质在色谱体系下发生互变
含活泼羰基化合物在特定的HPLC体系及溶剂中可能以原型、水合半缩醛/酮、醇合半缩醛/酮等多种形态存在,也有学者认为是酮式-烯醇式互变异构,其动态平衡受稀释剂种类、流动相pH值、柱温、存放时间等多重因素影响[1]。这种互变可能导致结构中生色团发生变化,使得不同形态化合物紫外响应存在差异。例如,乙酰乙酸乙酯酮式结构UV λmax为204nm和280nm且吸收系数ε很小,而烯醇式结构λmax=245nm且ε很大,两者呈现出完全不同的紫外吸收特征[2]。
此类化合物测定时可能产生多个色谱峰,且其在溶液和色谱体系中的存在形态将影响校正因子结果。例如头孢吡肟EP杂质C,其对照品溶液配制后峰面积迅速增大,而后趋于平衡,测得校正因子值与0h比较变化明显。
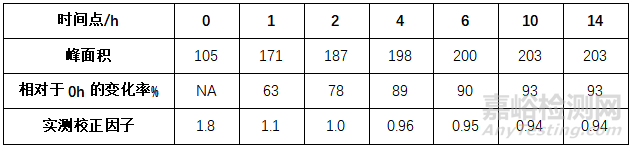
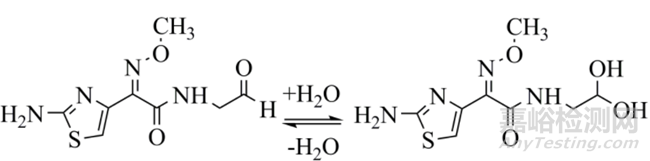
图 头孢吡肟EP杂质C原型与水合半缩醛互变
此类化合物校正因子测定结果的影响因素多,实测结果可能与药典差异非常大,例如某品种杂质实测值为1.5,而药典标准为3.1,可通过结构特征解释此种差异,并优选采用法定标准收载的校正因子值。
3.4 杂质结构错误
药典中收载的杂质结构或企业获得的标准物质结构可能存在错误,导致校正因子测定值存在差异。笔者曾遇到某品种杂质,其结构收载于USP中,按照此结构采购对照品后,实测RRT和校正因子均与USP相差甚远,后经LC-MS、DAD光谱等研究发现,USP中收载的杂质结构有误,遂在该品种申报时修正了杂质结构、RRT及校正因子。
另一个案例是,杂质对照品(供应商A)定位结果、校正因子与法定标准存在明显差异,于是从供应商B处采购了该杂质,结果显示两家供应商杂质RRT不同,且供应商B提供的对照品测得的RRT、校正因子与药典基本一致,后经对1H-NMR谱进行详细分析,并结合2D-NMR,发现供应商A提供的杂质对照品基团连接位置有误。
3.5 检测波长不合适
通常HPLC检测波长应尽量避开吸收值急剧变化波段,但由于各杂质的UV特征可能存在差异,有时无法兼顾所有杂质。当所选择检测波长在杂质吸收值急剧变化波段,而主成分则在缓慢变化波段时,由于HPLC检测器对于波长的准确度要求一般为±2nm,不同仪器间的波长最大可相差4nm,可能会对测得校正因子产生较大的影响。
此种情况,建议对检测波长附近的波长分别进行校正因子研究,结合药典收载值,基于严格控制杂质策略选择合适的校正因子,或改变检测波长,必要时可采用双波长法分别控制不同的杂质。
也有文献报道[3],不同的检测器可能影响校正因子值,佐匹克隆杂质D采用DAD和VWD检测器测定校正因子值分别为0.828和1.129,两者相差超过30%。
3.6 色谱柱选择不合适
色谱柱是HPLC分离的关键,可能影响保留时间和响应、分离度等,部分化合物可能与色谱柱填料中残留的硅醇基、金属离子等发生相互作用,进而影响校正因子测定结果。
有同行研究发现[4],某杂质采用三根不同色谱柱测得校正因子分别为4.1、2.3和1.6,法定值为1.3,推测化合物中的嘌呤结构可能与色谱柱中的硅醇基发生吸附作用而导致。降低流动相pH值可使得实测校正因子的重复性良好且与法定值一致。当然,也可选用封端色谱柱,并在质量标准中注明。
因此,当选用USP或EP标准,建议尽量选择该品种官方推荐的色谱柱开展研究,具体色谱柱信息可在EDQM或USP官网获得。
3.7 关注标准收载杂质及对照品是否含盐基
应关注药典收载杂质结构信息,明确杂质是否含盐基。例如,盐酸丙米嗪EP杂质A(去甲丙米嗪),ChP2020质量标准及中检院对照品为盐酸盐,而EP质量标准则不含盐基;瑞舒伐他汀钙USP相关杂质A和相关杂质B对照品均为钙盐,而质量标准则为酸形态。
因此,应根据质量标准中杂质和对照品的存在形态,在计算时予以考虑、折算,尤其是当对照品为柠檬酸盐、二乙胺盐、四甲基铵盐等分子量较大的盐基形态时,可能严重影响校正因子测定结果。
3.8 标准曲线测定截距过大或浓度范围不合适
单点法计算校正因子的前提是线性良好且假设标准曲线经过原点,标准曲线法使用斜率计算校正因子的前提是假设截距为0,因此线性截距大小将影响校正因子结果。目前常规做法未将截距校正为0,有文献报道[5],当标准曲线相关系数不低于0.999且斜率与截距的比值大于100时,截距可忽略不计。
因此,应控制线性斜率在一定范围内。正的截距表示高浓度响应的饱和度或相应处有干扰存在,负的截距说明可能方法灵敏度存在问题或分析物质残留在容器或HPLC系统中[5]。同时,标准曲线的浓度范围亦影响校正因子结果,杂质和主成分的浓度水平应相当,涵盖定量限至限度以上,避免使用过高的浓度影响线性斜率。
四、总结
2012年CDE化药共性问题解答中提到:考虑到校正因子测定的影响因素,如所用分析方法中相关条件均一致,可以直接采用ChP、USP、EP/BP或者其他权威公开标准中的相应杂质的校正因子;如相关条件发生变化,则需要重新测定。
由于校正因子影响因素多,同一品种采用相同方法不同,不同药典收载的校正因子可能也不同,例如头孢吡肟杂质E,USP和EP校正因子分别为2.1和1.8,因此一般应进行校正因子验证,并重点关注校正因子的重复性和耐用性。当校正因子与药典差异较大时,可从对照品、分析方法、操作过程、药典的准确性等方面进行分析,如实测结果确实与药典差异较大,因在申报资料中予以说明。
参考文献:
[1] G. Liu, B. Luan, L. Xin, et al. J. Chromatogr. A, 2019, 1594: 112-119.
[2] 沈建红, 秦向东, 裴静, 等. 现代仪器, 2002, 6: 14-15.
[3] 洪淑华, 林琪珊, 柴将红, 等. 中国药物评价, 2023, 40(5): 384-389.
[4] 公众号:小花晒太阳. 校正因子的实测值与法定值不一致.
[5] 肖亭, 王晨, 姚尚辰, 等. 药学学报, 2020, 55(12): 2854-2861.

来源:注册圈


