您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-07-27 11:26
摘 要 / Abstract
作为超出《与贸易有关的知识产权协议》(TRIPS)条款,生物制品试验数据保护制度已成为近年来国际多边或双边贸易协定和国内产业政策制定的热点。通过回溯制度根源、比较制度价值发现,一方面,创新生物制品的试验数据对生物类似药产生了正外部性,即允许后者依赖前者的试验数据,仅证明其与前者相似即可获批上市,这成为该项制度产生的根源。另一方面,从比较角度,作为规制外部性的手段,试验数据保护已成为专利保护的有益补充,且在生物制品领域尤为明显,诸如在保护对象、保护力度、实质保护期限等方面。基于上述发现,建议我国未来在制度建构时更好地平衡“创新激励”与“药品可及”这一知识产权保护的原始命题,结合我国国情设置相匹配的制度强度,同时需要注重与生物制品注册分类及审批制度、专利保护制度的协同与衔接。
The biologics test data protection system, as a TRIPS-plus provision, has become a focal point in international multilateral or bilateral trade agreements and domestic industrial policy formulation in recent years. This paper examines the origin and value of this system through a retrospective analysis and comparative evaluation. It finds that the test data of innovative biologics generate positive externalities for biosimilars by allowing them to rely on the former’s data, thereby only needing to prove similarity for market approve. This is the root cause of the system. From a comparative perspective, test data protection complements patent protection, especially in the biologics field, in terms of protection object, intensity, substantive protection period, etc. Based on these findings, this paper suggests that China needs to balance the original proposition of intellectual property protection between "innovation incentive" and "drug accessibility" in future system construction, tailored to China's national conditions. At the same time, it is necessary to pay attention to the coordination and connection with the registration, classification, approval systems and patent protection of biologics in order to construct a biologics test data protection system with Chinese characteristics.
关 键 词 / Key words
生物制品 ;试验数据保护 ;制度根源 ;制度价值 ;专利保护
biologics; test data protection; system origin; system value; patent protection
药品试验数据保护是一项由《与贸易有关的知识产权协议》(Agreement on Trade-related Aspects of Intellectual Property Rights,TRIPS)引入全球知识产权框架的制度,旨在保护创新药研发过程中的试验数据[1]。传统概念上的药品试验数据保护主要针对化学药品范畴,如美国1984年通过《药品价格竞争与专利期补偿法案》(Drug Price Competition and Patent Term Restoration Act)建立的药品试验数据保护针对的是新化学成分药品(new chemical entity,NCE);TRIPS协议第39.3条规定的保护对象亦为NCE。近年来,随着生物制品产业的迅速发展,美国、欧盟等国家和地区以及国际双边或多边贸易协议已逐渐将试验数据保护范围扩展至生物制品[2],生物制品试验数据保护成为后TRIPS时代该项制度发展的新趋势。
为了遵循TRIPS协议的知识产权保护标准,我国曾于2002年通过《药品管理法实施条例》进行了国内法转化,规定为含有新型化学成分药品提供6年的数据保护期。但由于缺乏可操作性配套法规,该项制度未予落地实践。值得关注的是,近年来国际背景与国内环境方面均发生了新的变化。一方面,在国际贸易对话与谈判中,生物制品试验数据保护逐渐成为发达国家和地区关注的知识产权保护焦点之一,我国亦在2014年7月1日生效的《中华人民共和国和瑞士联邦自由贸易协定》中包含了生物制品数据保护条款[3];另一方面,国内法律建设也已考虑将生物制品纳入试验数据保护制度中。例如,2017年10月,中共中央办公厅、国务院办公厅印发的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》提出,要完善和落实药品试验数据保护制度,对包括创新治疗用生物制品在内的相关药品给予一定的数据保护期[4]。2018年4月,国家药品监督管理局发布《药品试验数据保护实施办法(暂行)(征求意见稿)》,提出给予创新治疗用生物制品12年数据保护期[5]。2022年5月,国家药品监督管理局发布《药品管理法实施条例(修订草案征求意见稿)》,提出“国家对获批上市部分药品的未披露试验数据和其他数据实施保护”,保护期限6年[6]。截至目前,上述两项征求意见稿尚未正式发布。
总之,不论是源于自身制度需求,还是受于国际协定与国内政策的约束,我国生物制品试验数据保护制度的建立势在必行,战略焦点是如何将该项制度做好本土化移植。因此,从制度本质出发,探寻其在知识产权保护体系中的价值,将有益于我国未来制度方案的设计与完善。
1、制度根源:试验数据的外部性
(一)生物制品试验数据保护制度内涵
生物制品通常是指以微生物、细胞、动物或人源组织和体液等为起始原材料,用生物学技术制成,用于预防、治疗和诊断人类疾病的制剂[7]。根据产品创新属性,生物制品可进一步细分为创新生物制品(new biologics)和生物类似药(biosimilars)。创新生物制品通常包含新的活性成分,在研发过程中需要开展完整的临床前研究、临床研究以证明安全性、有效性和质量可控性。生物类似药是指在质量、安全性和有效性方面与已获准注册的参照药具有相似性的治疗用生物制品[8]。生物类似药大多是以创新生物制品为参照药而进行的仿制产品,但是由于生物学特性不同,其无法像化学仿制药做到与参照药完全一致,而只能达到“相似(similar)”,因此业界通常将生物制品的仿制药称为生物类似药。
生物制品试验数据保护制度的本质是对申请人在新药临床试验申请和上市申请时提交的付出相当努力后所取得的未经披露的试验数据或其他数据给予保护,以免不正当商业使用[9]。从执行层面来看,该制度在创新生物制品上市后赋予一定期限的保护期,在保护期内药品监管部门要保护该数据以防被泄露,并不受理和(或)不批准未经许可使用该数据的生物类似药上市,从而赋予创新生物制品市场独占权;数据保护期届满后,生物类似药便可依赖其试验数据进行上市审批,从而寻求“创新激励”与“药品可及”之间的利益平衡。
(二)试验数据的外部性
一方面,试验数据的重要性与巨额投入。药品的特殊性使其在上市前必须经过药品监管部门的审查,证明其具有安全性、有效性和质量可控性。因此,新药研发呈现出高投入、高风险、长周期的特点。《自然》杂志(Nature)公布的数据显示,2014年新药平均研发成本约为26亿美元,包含了失败成本和机会成本,平均耗时14年[10]。鉴于生物制品在理化特性、生物活性等方面的特殊性,创新生物制品研发面临更高的资金成本和时间成本。另有研究表明,2012~2014年创新生物制品临床阶段的开发成功率仅为18%[11],这更加印证了创新生物制品研发高风险的特点。综上,创新生物制品研发过程所获得的数据是研发者付出创造性劳动的成果,是新药上市审批的基本依据。而在获取上述数据的研发过程中,相较于化学创新药,创新生物制品投入的时间成本、风险成本更大,需要在上市后予以补偿。
另一方面,试验数据的外部效应。在经济学领域,外部性是指在缺乏任何相关交易的情况下,因单位或个人的生产或消费行为对他人或社会所产生的影响,这种影响有好有坏、有利有弊,进而又可分为“正外部性”和“负外部性”。例如,生产化工产品带来的环境污染,对于整个社会即为负外部性;发明人的发明创造带来了科学技术的发展,对于整个社会即为正外部性[12]。
生物制品试验数据的外部性体现为两点:其一,试验数据是满足创新生物制品安全性、有效性、质量可控性等上市审批要求的根本,而据此上市的创新生物制品将提高社会公众对新药的可获得性,尤其可以满足那些尚无有效治疗药物的患者需求,显著提高公众健康水平。其二,许多国家和地区在药品注册审批制度中均建立了生物类似药的简化申请程序,允许其依赖创新药的试验数据,仅证明其与参照药相似即可获批上市。这种情况下,生物类似药方不仅避免了巨额投入,而且可以实现快速上市。可见,不论是对仿制药方还是社会公众,生物制品试验数据均带来了积极效益,体现为正外部效应。
(三)试验数据保护制度对外部性的法律规制
从经济学角度看,外部性直接导致了私人成本与社会成本之间的差异,造成资源配置效率低下,是市场失灵的重要表现;从法学角度看,外部性容易在社会群体之间造成权利冲突、利益失衡,进而影响社会的稳定与和谐[13]。外部性问题的存在必然刺激政府通过法律进行干预和规制,从而通过制度的矫正,使私人收益与社会收益之间达到有效平衡。
生物制品试验数据这种正外部性也可能使社会收益大于私人收益。在简化申请程序下,生物类似药可依赖创新药的试验数据,简化试验环节和上市注册的申报资料,极大减少研发成本与时间。例如,美国生物类似药上市只需提交资料证明拟上市药品的活性成分、给药途径、剂型以及生物等效性等与仿制对象相似,即可获批上市,而得以简化的动物实验、临床试验、分析研究等正是创新药研发成本耗费最大的部分(图1)[14]。这种简化申请程序使得创新药试验数据所蕴含的技术和知识很快被生物类似药模仿和依赖,产生不合理的“搭便车”行为,打击了创新药研发的积极性。如果这种损害不能获得很好的救济,那么长期来看,创新药研发的动力不足,社会将陷入创新枯竭的被动局面,从而整体上掣肘社会的发展。试验数据保护制度由此产生,一方面,赋予创新药一定期限的市场独占权以保护试验数据不被依赖;另一方面,在市场独占期结束后,允许生物类似药依赖创新药的试验数据,简化申请而上市,以更小的成本创造相同的社会价值,从而在“创新激励”和“药品可及”之间达成有效平衡。

2、制度价值:试验数据保护制度与专利制度的比较
知识产权制度致力于平衡“创造”与“普及”的固有矛盾。作为主流知识产权保护形式,专利制度亦可通过赋予新药市场独占权发挥规制试验数据外部性的作用。那么相较之下,试验数据保护制度价值如何?对这一问题的分析将有助于完善我国制度设计,更好发挥二者协同作用。
(一)试验数据保护制度与专利制度的区别
作为知识产权保护形式,专利制度和试验数据保护制度在制度效果上较为相似,均可为权利人相关产品带来一定期限的市场独占权。但是从制度内容来看,二者则存在较多区别(表1)。其一,权利授予体系不同。专利授权与药品审批属于两个独立体系。专利由知识产权部门按照“申请-公开-审查-授权”程序,重点审查其相较于现有技术有无新颖性、创造性和实用性,技术要求较为严格。试验数据保护制度与药品注册程序密切相关,其权利的获得是在新药获批上市后,通过药品监管部门授予的一项市场独占权,具有一定的行政保护色彩[15]。其二,保护对象不同。专利制度保护的是具有新颖性、创造性和实用性的研究成果,是药品研发过程中发现的新的药物活性成分、制备方法、药物用途等技术方案,因此一个上市药品可能存在多项专利,一项专利也会涉及多个上市药品。而试验数据保护制度保护的则是创新药研发过程中用于证明药物安全有效的试验数据和其他数据,其权利赋予获批的上市药品,因此其与上市药品具有严格的一一对应关系。其三,权利内容与实现方式不同。专利权具有独占实施、许可、转让等民事财产权,试验数据保护则拥有数据独占权,二者均属于对世权,但是在权利实现方式上存在差异。专利保护针对的是已上市仿制药涉嫌侵权的行为,通常是通过司法程序,由法院判决制裁专利侵权人而实现的一种司法保护方式。而试验数据保护针对的是申请上市的仿制药(尚未上市),主要通过药品监管部门不受理和(或)不批准仿制药申请的行政决定而实现。其四,保护期限不同。专利保护期通常为20年,自专利申请日起算;生物制品试验数据保护期相对较短,5~12年不等,如美国规定12年、欧盟10年、日本8年、土耳其6年、澳大利亚和加拿大均5年等[16],但均从创新药获批之日起算。

(二)试验数据保护制度是对专利制度的补充
作为主流知识产权保护形式,专利制度在激励创新、维护创新成果利益方面的价值不可替代。而试验数据保护制度直接嵌入药品注册审批程序,与上市药品形成一一对应关系,成为专利制度的一项重要补充,尤其是在生物制品领域。
1.保护对象:弥补专利制度的不足
一方面,药品专利制度侧重于对研制或分离出的化合物、氨基酸序列以及相关方法等技术方案进行保护,且需要符合新颖性、创造性和实用性的审查标准,而创新药研发过程产生的试验数据和其他数据虽然耗费了巨额投入与努力,但通常因不属于技术方案而不具备专利授权条件,因此试验数据保护制度的建立恰好弥补了对这类劳动成果的保护缺位。另一方面,从市场独占结果来看,虽然大多数创新生物制品可以得到专利保护,但不能排除有一部分首次上市的创新生物制品从市场角度而言属于新药,但由于活性物质早已存在等原因不符合新颖性、创造性和实用性标准而无法得到有效的专利保护,此时试验数据保护成为有效补充。
值得关注的是,生物制品对这一有效补充的依赖更为明显。与化学药品相比,生物制品分子量大、结构复杂,在理化性质、生物活性、质量控制和免疫原性等方面存在显著区别,因此往往无法像化学药品一样采用马库什结构获得较宽的专利保护范围,通常在专利的申请和审查过程中,其专利保护范围最终缩小至某个特定的蛋白质序列,或与该特定序列高度相似的一系列序列,保护范围较窄。加之,正是由于生物制品技术属性的特点,不同于化学仿制药须与参照药达到“一致性”的审批标准,当前许多国家和地区对生物类似药上市审批仅要求其与参照药“相似”即可,且允许在生产工艺方面与参照药有相对更大的偏差,这就增加了生物类似药绕开专利保护范围进行开发的可能。这种回避专利的现象称为生物制品领域的专利保护空白(patent protection gap)[17]。因此,相较于化学药品,生物制品更依赖于试验数据保护制度。
2.保护力度:试验数据保护的稳定性更强
专利保护针对的是已经上市的生物类似药涉嫌侵权的行为,是通过司法程序,由法院判决制裁专利侵权人而实现的一种司法保护方式,是与药品注册审批平行的体系。如图2所示,由于目前我国生物制品专利链接制度有待进一步完善,当创新生物制品获批上市后,即便处于专利保护期,生物类似药仍可以随时提交上市申请,在满足注册要求后即可获得药品监管部门批准上市。此时,作为专利权人的创新生物制品只能通过提起侵权诉讼予以维权,即专利保护无法完全阻断生物类似药冒险上市途径。
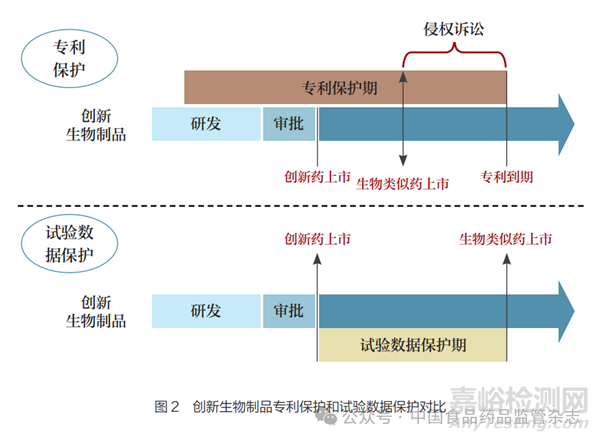
相较于专利保护,试验数据保护的授权和执行直接嵌套于生物制品注册审批过程中。当生物类似药在创新生物制品获批上市后提交上市申请时,药品监管部门即会核查其参照药品的数据保护情况,只有当其数据保护到期后,生物类似药方可获批上市。因此,从创新药市场独占权的预期来看,试验数据保护实现的市场独占权更为稳固。
3.保护期限:弱专利保护状态的有效补充
理论上,专利保护期远长于试验数据保护期。然而对于创新药而言,其核心专利申请往往在创新药研发最早期就开始,虽然长达20年,但在此期间要经历漫长的临床试验和上市审批过程,使得其在创新药上市后剩余的有效专利期明显缩短,部分创新药上市后甚至难以实现盈亏平衡[18]。而试验数据保护则直接从创新药获批之日起算,不存在实质的市场独占期缩短的问题,因此即使5~12年试验数据保护期看起来比20年专利保护期短,但实际有效保护期反而有可能比专利保护期长,尤其是在弱专利保护状态下。Grabowski等[17]曾研究在不同专利保护强度下试验数据保护制度对创新生物制品盈亏平衡的影响,指出在弱专利保护状态下,即创新药上市后有效专利保护期平均仅为7年的情况下,如果仅有专利保护,创新药在上市后25年时仅有14%的可能性达到盈亏平衡,而赋予其12年的试验数据保护期后,这一概率显著提高至62%,表现出较强的激励作用,表明试验数据保护是对专利保护的有效补充。
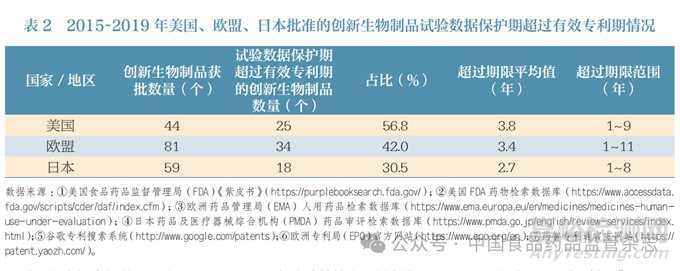
为了更好地验证这一补充作用,本文统计了2015~2019年美国、欧盟、日本批准的创新生物制品试验数据保护期和上市后有效专利期的重叠情况,其中纳入统计的专利为创新药的物质专利。经统计,美国、欧盟和日本均存在试验数据保护期超过上市后有效专利期的情况,占比分别为56.8%、42.0%、30.5%,超出期限的平均值在2.7~3.8年之间(表2)。可见,试验数据保护确实发挥了有效补充专利保护的作用,尤其是对于创新生物制品数据保护期限较长的国家和地区。例如在美国,即便考虑到药品专利期延长的因素,创新生物制品自获批之日起12年的试验数据保护期仍具有长周期优势。
3、制度建构:对中国方案的几点建议
(一)立足“创新激励”与“药品可及”的有效平衡
作为一项由TRIPS协议引入的新型知识产权,药品试验数据保护制度的实施同样具有双面性。一方面,该制度赋予创新药一定的市场独占期,发挥激励创新作用;另一方面,该市场独占期会不可避免地造成创新药市场垄断、价格高昂,同时延缓生物类似药的上市进程,影响社会公众对于药品的可及性[19]。这种影响的程度主要取决于制度强度与本国国情的匹配水平。
《2023年度药品审评报告》显示,2023年我国受理创新治疗用生物制品新药上市许可申请45件(29个品种),同比增加136.84%;建议批准创新治疗用生物制品新药上市许可申请19件(15个品种),同比增加111.11%[20]。可见,近年来随着生物医药企业创新投入的持续增加与生物制品创新公司的不断涌现,我国创新生物制品产出已初现成果,未来仍须保持政策激励。与此同时,从药品可支付的角度来看,我国人均可支配收入水平在全球范围仍处于较低水平,社会公众可承受的药品支出水平和社会医疗保险基金的总体承受能力仍有限。因此,建议在设计生物制品试验数据保护制度强度时立足“创新激励”和“药品可及”的有效平衡,制度强度多以保护期限、保护方式等核心机制来体现。
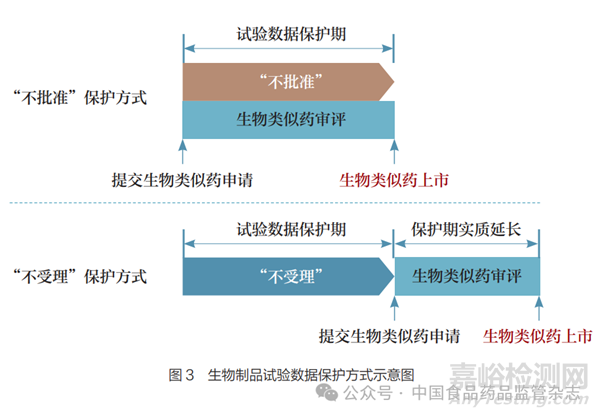
以保护方式为例。除了TRIPS协议要求的“不披露”“不依赖”以外,国际通行做法还包括“不受理”“不批准”或者两者的结合使用。例如,美国的创新生物制品试验数据保护期为12年,其中前4年“不受理”,后8年“不批准”。“不受理”和“不批准”两种保护方式体现了不同的制度强度(图3)。“不受理”能够实质延长试验数据保护期,因为在保护期届满后生物类似药方可提交并开展审评,而这段时间无疑延长了创新生物制品的市场独占期。相比之下,“不批准”可以实现一旦保护期届满,生物类似药即刻上市。但“不批准”也存在一定的弊端,如可能导致生物类似药申请提交节点过于散乱,造成不必要的审评压力。因此,建议我国在制度建设中选择“不受理+不批准”分段保护方式。
(二)衔接生物制品注册分类与审批程序
与专利权等其他知识产权不同,试验数据保护的实施与生物制品注册密切相关,其权利授予形成于创新生物制品注册审批过程,其权利实施体现于生物类似药注册审批过程。可见,虽然试验数据保护的权利根源于试验数据的外部性,但为了便于操作,在制度实然层面体现为将权利授予试验数据支撑的药品申请上。因此,在制度设计时需要特别注意与我国生物制品注册体系的衔接,具体包括保护对象与生物制品注册分类的衔接、权利实施与生物类似药审批程序的衔接。
根据国家药监局于2020年6月发布的《生物制品注册分类及申报资料要求》,我国将生物制品分为预防用生物制品、治疗用生物制品和按生物制品管理的体外诊断试剂三大类,其中治疗用生物制品可细分为1类创新型生物制品、2类改良型生物制品、3类境内或境外已上市生物制品(含生物类似药)[7]。而预防用生物制品和按生物制品管理的体外诊断试剂并无“生物类似药”的注册类别。由此,按照权利授予和权利实施衔接的角度,无依赖即无保护,建议我国未来生物制品试验数据保护对象限于治疗用生物制品。此外,除了对1类创新型生物制品给予保护外,2类改良型生物制品以及3类境外已上市生物制品(但在中国首次上市)从试验数据外部性角度来看也可能产生相应的被依赖的可能,因此我国是否亦将其纳入保护,需要结合具体实际谨慎设计。
(三)协同专利保护制度
如前所述,从对创新生物制品知识产权保护结果来看,生物制品试验数据保护与专利保护基本一致,即均能赋予创新药一定期限的市场独占权,但目前我国生物制品专利链接制度有待进一步完善,专利保护和药品审批尚无链接,因此在赋予市场独占权的稳定性或直接性方面,试验数据保护制度更有优势。同时,相较于化学药品,生物制品领域存在“专利保护空白”现象,使得其更依赖试验数据保护制度。因此,建议我国从激励创新的整体政策效果出发,通过具体案例分析和数据测算,在掌握并考虑生物制品专利保护实践情况的基础上,设计试验数据保护制度,以期更好地发挥二者的协同作用。

来源:中国食品药品监管杂志


