您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-08-03 09:31
摘 要 / Abstract
目的:根据中美同情用药经验,调研临床试验从业人员的认知和理解现状,对细化和完善我国同情用药制度体系提出建议,从而更好地满足患者的临床需求。方法:使用自愿抽样、方便抽样等非概率抽样方法,基于现有中美同情用药经验,就同情用药的适用条件、申请和审查主体、患者安全管理、审批流程等方面,对临床试验从业人员的认知和理解进行问卷调查。结果:共收集740份有效问卷。调研结果显示,我国临床试验从业人员对我国同情用药的认知程度有待提高。相关企业和机构人员对我国同情用药的认知与理解存在共性和差异。共性主要体现在对同情用药适用人群、“是否可以向药品监管部门提出同情用药申请”的审查主体、严重不良事件报告义务的承担方、同情用药开展地点以及收费问题的认知等方面。差异主要体现在同情用药的药品条件、申请主体、同情用药期间发生不良事件的补偿或赔偿承担方、美国食品药品监督管理局针对紧急情况下单个患者协议和非紧急情况下中等规模患者同情用药的审批流程等方面。结论:现阶段,我国临床试验从业人员对同情用药的了解不足。不同角色的临床试验从业人员对我国同情用药的药品条件、申请和审查主体、患者安全管理以及审批流程等方面的理解差异较大。因此,鼓励加强同情用药相关政策宣传工作,引导更多满足条件的企业开展同情用药计划,让更多潜在患者获益,同时建议针对理解差异较大的内容进一步明确、细化和完善。
Objective:To explore the cognitive status and understanding of professionals related to clinical trials based on compassionate use experiences in China and the United States.It provides suggestions for refining and improving China’s compassionate use system to better meet patients' clinical needs.Methods:Non-probability sampling methods,such as voluntary and convenience sampling,are used.A questionnaire survey is conducted with professionals related to clinical trials,focusing on their cognition and understanding of the applicable conditions of compassionate use,the application and review subjects,patient safety management,and the approval process.Results:A total of 740 valid questionnaires are collected.Survey results shows that the awareness of Chinese clinical trial practitioners on compassionate use in China needs to be improved.There are commonalities and differences in cognition and understanding between enterprise practitioners and institutional practitioners.Commonalities are mainly reflected in their cognition of the population suitable for compassionate use,the review subject of applications for compassionate use to the National Medical Products Administration,parties responsible for patient adverse event reporting,places for compassionate use,and fee issues.Differences exist in drug conditions,application subjects,compensation for adverse events during compassionate use,and the FDA’s approval process for compassionate use of individual patients in emergency situations and medium-sized patients in non-emergency situations.Conclusion:Clinical trial practitioners in China have a significant lack of understanding of the country's compassionate use system.Survey results suggest that clinical trial practitioners in different roles have varying levels of understanding of drug conditions,applicants,patient safety management,and approval processes for compassionate use in China.Further policy advocacy work is recommended to guide more qualified enterprises to carry out compassionate use programs to benefit more potential patients.In addition,the content of understanding that shows significant differences needs further clarification,refinement,and improvement.
关 键 词 / Key words
同情用药;拓展性临床试验;认知现状;适用条件;申请和审查主体;安全管理;审批流程
compassionate use; extended clinical trials; cognition; applicable conditions; application and review subjects; safety management; approval process
拓展性临床试验有时也称同情用药[1]。《药品管理法》第二十三条涉及相关描述:对正在开展临床试验的用于治疗严重危及生命且尚无有效治疗手段的疾病的药物,经医学观察可能获益,并且符合伦理原则的,经审查、知情同意后可以在开展临床试验的机构内用于其他病情相同的患者[2]。
美国作为全球最早建立同情用药制度的国家,自20 世纪70 年代开始致力于推行该制度。1987 年,美国食品药品监督管理临床研究审批机制[3]。1997 年,美国《食品药品管理现代化法案》(Food and Drug Administration Modernization Act)对单个患者、中等规模患者、大规模患者的同情用药均作出明确规定[4]。2016年, 美国国会通过的《21 世纪治愈法案》(21st Century Cures Act)要求药物研发企业在公共网站发布同情用药的实施计划[5]。FDA 在同情用药方面对企业、医生、患者各方需要了解的信息,以及各自的责任与义务都作出详细规定[6]。
我国对同情用药的探索相对较晚。2017 年,《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》中首次提及了拓展性临床试验的概念[7]。同年,原国家食品药品监督管理总局药品审评中心组织起草了《拓展性同情使用临床试验用药物管理办法(征求意见稿)》(以下简称《同情用药征求意见稿》),就拓展性同情使用临床试验用药物的使用情形、基本要求、申请与审批等征求意见[8]。2019 年,《药品管理法》第二十三条再次提及同情用药相关内容[2]。
在我国药品监管历程中,同情用药的案例较少[9]。目前,我国已系统探索了同情用药的定义、使用情形、基本要求、申请与审批,具体细节仍有待细化,同时安全管理相关内容仍需补充。本文针对上述情况,结合中美同情用药经验设计调查问卷,主要从同情用药的适用条件[10](包括人群及药品)、申请和审查主体(包括申请[11] 及伦理审查[12-13])、安全管理(包括不良事件的记录报告义务以及补偿或赔偿问题[14])、审批流程以及其他方面(如是否应当收费[15] 以及是否应当作为上市审批考虑因素[16-18] 等),对临床试验相关人员进行问卷调查,收集被调查人员基于现有认知对同情用药的理解差异情况,旨在为细化和完善我国同情用药制度体系提供建议。
1、材料与方法
(一)调研方法
本研究使用自愿抽样、方便抽样等非概率抽样方法选取调查对象进行问卷调查。样本量的计算可以在一定程度上参照概率抽样计算方法, 应用Cochran(1977)公式确定样本量。假设置信区间为99%(t=2.58),可接受的允许误差(d)为5%,并假设方差为0.25,以获得最大样本量。经过计算,得出的样本大小为663[19]。
Cochran(1977)公式:
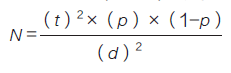
式中,N 为样本量,t 为标准正态分布的临界值,p 为总体比例,d 为可接受的允许误差。
(二)调研对象
同情用药又称拓展性临床试验,为保证研究的有效性,本文选取的调研对象为对临床试验具有基本认知的临床试验相关从业人员(主要为企业以及临床试验机构人员)。其中,企业人员主要包括合同研究组织的临床监查员或临床协调员,此外还包括药物警戒专员、项目经理、质量控制专员以及制药企业研发人员等。临床试验机构人员主要包括研究者、临床试验机构管理办公室及医院伦理委员会人员以及护士。
(三)研究方法与内容
本研究于2022 年2 月28 日至2022 年6 月30 日期间以问卷星和药研社为媒介开展问卷调查,被调查者在线提交电子问卷,数据存储在相关平台。本研究围绕被调查者对同情用药的基本认知以及同情用药的适用条件、申请和审查主体、患者安全管理、不同规模人群的审批流程等方面展开,收集并分析不同从业人员对上述各方面的认知和理解差异。
(四)数据处理与分析
本研究采用IBM SPSS Statistics 22 软件进行统计学差异性分析。根据SPSS 分析规则,若频数大于5,采用卡方检验;若频数小于5,采用费希尔(Fisher)精确检验。
2、结 果
(一)基本概况
本研究共收集调查问卷749份,其中有效问卷740 份,答复率约为98.80%,符合最小样本量要求。被调查者中,企业人员有437 人(59.05%), 其中含424 名合同研究组织从业人员,13 名制药企业人员;临床试验机构人员有303 名(40.95%)。
(二)调研结果
1. 对同情用药的基本认知
740 名被调查者中, 仅有239 名(32.30%) 达到了解国内同情用药发展现状的水平。该数据在一定程度上反映出,我国临床试验从业人员整体对同情用药的关注度和参与度有待提高。其中,临床试验机构与企业从业人员对同情用药的认知水平存在显著性差异,前者的总体了解程度较高(P<0.001,图1)。
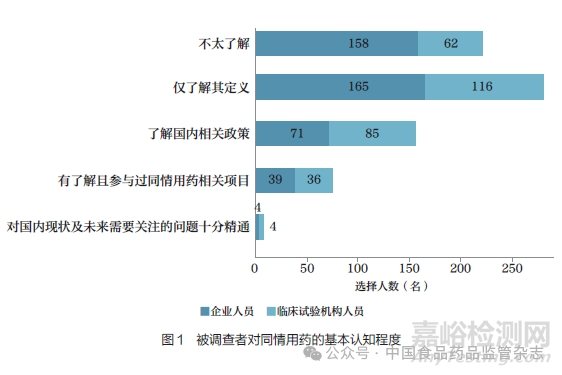
2. 同情用药的适用条件
结果显示,临床试验机构与企业的从业人员对同情用药适用人群的理解不存在显著性差异(P>0.05)。其中, 均有接近50% 的被调查者对于将“因不符合临床试验入组/ 排除标准”以及“因地域或时间限制等原因”无法参加新药注册临床试验的患者作为同情用药对象存在顾虑(图2)。
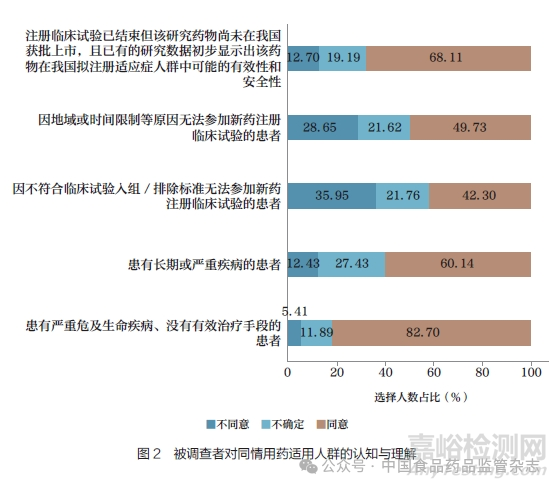
在同情用药的药品条件上,临床试验机构与企业从业人员的认知与理解存在显著性差异(P<0.05)。临床试验机构人员相比企业人员对国内药品临床试验开展阶段的要求更加严格,且更加注重其他国家和地区的相关研究结果;企业人员则更关注药品在国内后续的上市申请计划(图3)。
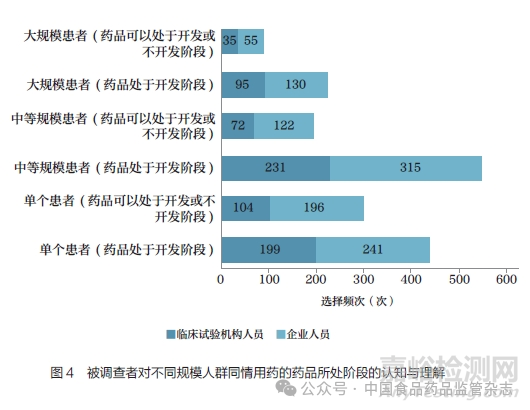
同时,调查问卷中依据美国同情用药经验将适用人群划分为单个患者、中等规模患者以及大规模患者。对于中等规模以及大规模患者同情用药,临床试验机构与企业从业人员对于药品是否必须处于开发阶段的认知不存在显著性差异(P>0.05);对于单个患者同情用药,临床试验机构人员相比企业人员对药品所处阶段的要求更加严格(P<0.05,图4)。
3. 同情用药申请和审查主体
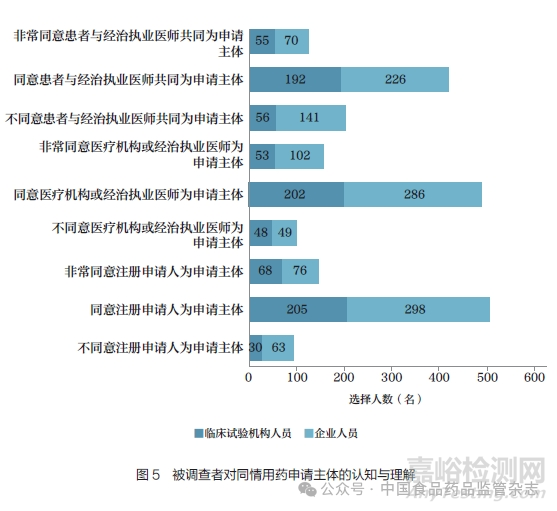
临床试验机构与企业从业人员对于是否将“注册申请人”以及“医疗机构或经治执业医师”纳入申请主体的认知无显著性差异(P>0.05), 但企业人员对于将患者纳入申请主体的顾虑较临床试验机构人员更大(P<0.01,图5)。此外,在同意或非常同意由注册申请人向药品监管部门提出申请的被调查者中,超过95%(636/647)的人认为医疗机构需要对此提供专业性意见。
关于“是否可以向药品监管部门提出同情用药申请”的审查主体,临床试验机构与企业从业人员的认知无显著性差异(P>0.05),双方对于经治执业医师及其所在临床试验机构伦理委员会作为审查主体的认同比例均超过50%(表1)。此外,少部分被调查者补充可以将患者本人或法定代理人、有经验的研究者和科室团队(行业关键意见领袖)、法律专家纳入审查团队。在选择临床试验机构伦理委员会作为审查主体的被调查者中,有170 位临床试验机构人员所在机构有多个伦理委员会小组,其中144 人认为单个患者同情用药申请需要通过伦理委员会小组全体审查,26 人认为通过一名或几名伦理委员会小组成员审查即可。

4. 同情用药患者安全管理
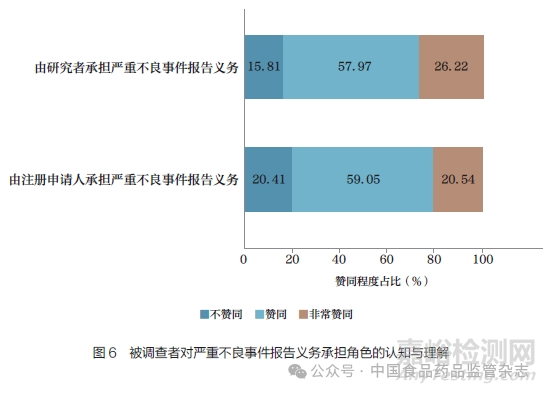
临床试验机构与企业从业人员对于“ 不良事件是否需要被记录” 的认知无显著性差异(P>0.05),双方均有超过95%的人认为在同情用药期间发生的不良事件需要或非常需要被记录。同时,双方对于严重不良事件报告义务承担角色的认知也无显著性差异(P>0.05,图6)。
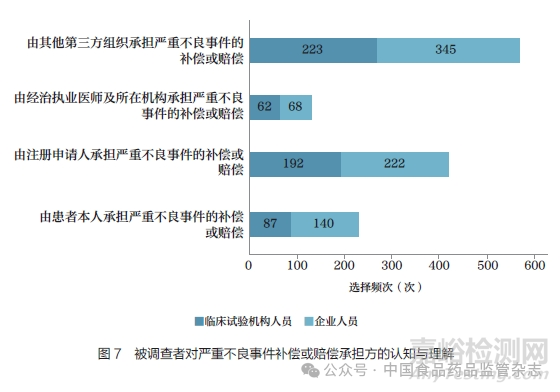
针对严重不良事件的补偿或赔偿问题,临床试验机构与企业从业人员对于承担方的认知差异较大(P<0.05),企业人员更倾向于选择患者本人或其他第三方组织,而临床试验机构人员更倾向于选择注册申请人或经治执业医师及其所在机构(图7)。另有调查者补充,补偿或赔偿的承担问题应考虑严重不良事件与药品之间的关系,以及临床试验申办方获益与否。
5. 不同规模患者同情用药审批流程
调查结果显示,对于单个患者、单个患者协议以及紧急情况下的单个患者同情用药审批流程,临床试验机构与企业从业人员的认知无显著性差异(P>0.05)。其中,对于单个患者同情用药,被调查者认为FDA 审批流程在我国不可行的原因主要为等待期太长。对于单个患者协议同情用药,认为FDA 审批流程在我国不可行的原因主要为伦理审查流程较慢。对于紧急情况下的单个患者以及单个患者协议同情用药, 认为FDA 审批流程在我国不可行的原因主要包括电话授权无法保留证据、患者用药不安全易出现医患纠纷等。此外,对于非紧急情况下的中等规模患者同情用药,超半数被调查者认为审批流程可以参照单个患者审批流程进行分类。而对于紧急情况下的单个患者协议以及非紧急情况下的中等规模患者同情用药的审批流程,临床试验机构与企业从业人员的认知具有显著性差异(P<0.05),企业人员认为FDA 审批流程不可行的占比更大(图8)。
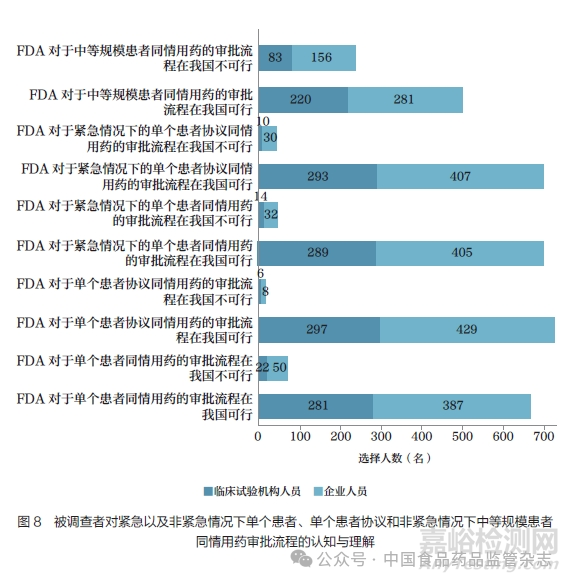
支持大规模患者同情用药的315 名被调查者中,临床试验机构与企业从业人员的认知无显著性差异(P>0.05), 双方均倾向于“在药物临床试验开始之前没有30 天的等待期,但在开始之前该方案必须得到药品监管部门的接收并获得临床试验机构伦理审查委员会的批准”。
6. 同情用药其他方面
临床试验机构与企业从业人员对于同情用药开展地点的认知无显著性差异(P>0.05),均有超过95% 的被调查者的认知与《药品管理法》第二十三条“经审查、知情同意后可以在开展临床试验的机构内用于其他病情相同的患者” 的规定保持一致[2]。
《同情用药征求意见稿》提出,“原则上不允许注册申请人对临床试验用药物收费”[8]。然而,关于“原则上”的定义尚不清晰。调查结果显示,临床试验机构与企业从业人员对于“是否应当收费”问题的理解不具有显著性差异(P> 0.05),并且与《同情用药征求意见稿》中的观点一致,少部分被调查者发表了对于规定中“原则上”的理解。
超过85% 的被调查者认为,同情用药数据应当成为药物上市审批的考虑因素。临床试验机构与企业从业人员对此问题的认知无显著性差异(P > 0.05)。
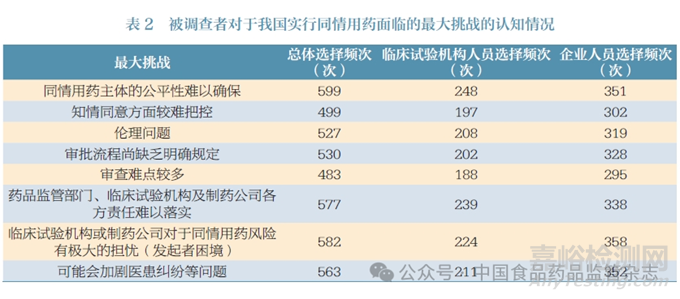
此外,调查问卷还列举了同情用药在我国可能面临的一系列挑战,由被调查者选择其认为的最大挑战。排名前4 的挑战依次为:同情用药主体的公平性难以确保;临床试验机构或制药公司对于同情用药风险有极大的担忧(发起者困境);药品监管部门、临床试验机构及制药公司各方责任难以落实;可能会加剧医患纠纷等问题。从被调查者担忧的侧重点来看,目前各方对于责任归属问题有较大顾虑(表2)。
(三)结果分析
对问卷调查结果进行研究分析发现,临床试验机构与企业从业人员对于同情用药的整体认知水平仍有待提高,其主要原因可能为对相关政策知晓度不足。通过加大政策宣传力度,增强临床试验机构与企业从业人员及患者对同情用药的了解,将有助于挖掘潜在同情用药计划,扩大受益人群。同时,由于临床试验机构与企业从业人员在同情用药临床试验中的角色不同,对于同情用药的认知与理解存在共性及差异。例如,双方对于同情用药适用人群的认知基本保持一致,对于“因不符合临床试验入组/ 排除标准”或“因地域或时间限制等原因”无法参加新药注册临床试验的患者持谨慎态度。同时,双方对于“是否可以向药品监管部门提出同情用药申请”的审查主体、严重不良事件报告义务的承担方等方面的认知不存在显著性差异。此外,双方对于同情用药开展地点以及收费问题的认知也不存在统计学差异,且与现有法规及《同情用药征求意见稿》中的内容保持一致。然而,在药品条件方面,企业人员更加关注后续的上市申请计划,而临床试验机构人员更加关注药品所处阶段以及既往研究结果。企业人员较临床试验机构人员更不同意将患者纳入同情用药申请主体的行列,且更加倾向于由第三方组织或患者本人承担同情用药期间严重不良事件的补偿或赔偿。对于紧急情况下的单个患者协议以及非紧急情况下的中等规模患者同情用药,企业人员认为FDA 审批流程不可行的占比更大。可见,临床试验机构与企业从业人员对同情用药期间患者安全和权益的考虑较为谨慎,企业人员对于患者安全责任归属问题的顾虑也可能在一定程度上限制其开展同情用药的积极性。
3、对我国开展同情用药研究的思考
本文基于调查结果中企业与临床试验机构从业人员对同情用药认知的共性及差异,分别针对参与各方工作的开展进行探讨,旨在满足共性要求的同时,充分考量认知差异化所反映出的问题,从而进一步细化和完善我国同情用药制度体系。
(一)监管部门
建议细化同情用药人群以及药品所具备的条件,进一步明确因不符合临床试验入组/ 排除标准或因地域/ 时间限制等原因无法参加新药注册临床试验的患者纳入同情用药目标人群的人群特征。同时,根据患者群体的疾病特征和治疗现状等,完善对不同规模同情用药研究的药品申请准入标准。
建议建立智能化线上提交及审批系统,明确相关材料提交要求以及审批流程,可以通过设立专门的同情用药部门等方式加快审批速度,缩短患者等待期,并明确患者等待期内是否可以进行其他药物治疗。
《同情用药征求意见稿》中虽提及“拓展性临床试验的研究数据一般不作为注册申请的主体资料”,但笔者认为,同情用药的结果仍不可被忽视。
(二)经治执业医师及临床试验机构
经治执业医师对患者病情更为了解,建议其与注册申请人共同作为同情用药申请人,帮助判断同情用药的必要性。
申请发起后,应由经治执业医师所在机构伦理委员会小组全体成员对申请的合理性以及同情用药计划进行审查,伦理委员会应建立健全快速的审查机制。
经治执业医师作为患者信息的首要来源,发现严重不良事件时应及时通知注册申请人以及伦理委员会,注册申请人应将相关情况及时上报至药品监管部门。
此外,通过建立并完善去中心化临床试验(decentralized clinical trial,DCT)模式,与患者所在偏远地区的临床试验机构建立医师网络与信息共享机制,从而为偏远地区患者同情用药提供安全保障。
(三)注册申请人
建议注册申请人在实施同情用药计划前为患者购买保险,将其他第三方组织(如基金会、保险公司等)作为患者补偿或赔偿的部分来源。同时,依据不良事件与药物之间的关系以及各方获益等因素,明确规定补偿或赔偿的情形、比例以及具体发放计划等内容。
此外,建议综合考虑药品的数据需求、申办方的研发生产成本、患者的经济情况、治疗周期以及患者获益情况,明确药品是否收费以及收费比例。
4、结 论
现阶段,我国临床试验相关从业人员对同情用药的认知程度有待提高。建议进一步加强同情用药相关政策宣传工作,引导更多满足条件的企业开展同情用药计划,让更多潜在患者群体获益。调研结果提示,我国同情用药参与各方在同情用药的药品条件、申请主体、患者安全管理、审批流程等方面的理解存在差异,相关制度规定需要进一步明确、细化和完善。
引用本文
向瑾,周姚,张庆*.基于对同情用药认知现状的调研探索在我国开展同情用药研究的思考[J].中国食品药品监管,2024(4):90-99.

来源:中国食品药品监管杂志


