您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-08-11 19:31
摘要
随着制药行业研发能力的下降,传统的临床试验终点在反映疾病状态变化方面的局限性日益凸显。数字健康技术通过提供连续、客观的健康数据,能够改善临床试验设计,提高治疗方案的个性化,并提升患者满意度。本文通过介绍杜氏肌营养不良症的步幅速度第95 百分位数和多发性硬化症的智能手机应用程序、特应性皮炎患者的夜间抓痒和睡眠监测以及癌性恶病质患者身体活动的测量等案例,展示了数字临床试验终点在药物开发中的应用,分析了我国数字临床试验终点的发展现状,并探索性地提出了加强监管科学研究和建立数字临床试验终点认证程序的建议,以期为促进我国数字健康技术驱动下的药物临床试验的发展提供借鉴。
关键词:数字健康技术;数字生物标志物;数字临床试验终点;临床试验研发;监管科学
制药行业正面临着研发能力下滑的挑战,2016 年制药行业的投资回报率(returnon investment,ROI) 降至历史低点3.7%。美国经济学家Scannell 称之为反摩尔定律(Eroom's law), 并指出尽管生物医学取得进步,但药物研发却变得更加缓慢,且成本高昂[1]。一个关键问题是临床研发阶段中“晚期失败”的增加,其失败的主要原因是未达到主要疗效终点或出现安全问题。有研究显示,1998~2015 年,640 种新疗法中有57% 因疗效不足、未能达到研究终点而失败[2]。麻省理工学院最近的一项研究是利用2 个大型数据集,发现了新药Ⅲ期临床研究的成功率仅为40%[3]。在药物研发过程中通常要投入大量资金,晚期失败的高频发生不仅会降低管线价值和组合价值,也会大幅度减少整体的研发ROI。
反摩尔定律还能反映出一个更深层次的问题, 即当前使用的临床试验终点未能捕捉到对疾病状态有意义的变化。那么数字临床试验终点能否有助于减少晚期失败的频率, 提升新药研发成功率?
1 以患者为中心的数字临床试验终点
1.1 现有临床试验终点的局限性
临床试验终点通常难以全面反映患者所承受的疾病负担。例如,死亡率作为重要的临床试验终点之一,可能仅揭示疾病影响的一个侧面。特别是对于那些本质上较为良性的疾病,其死亡率可能相对较低,此时选择死亡率作为临床试验终点则不太合适。但同时,这类疾病可能会通过诱发严重躯体症状、影响生理状态而极大地影响患者的生活质量。
目前,临床试验终点设计通常采取间歇性的评估方法,这意味着研究者只能在特定时间点对患者进行一次性的快照式评估,而在两次评估之间可能存在许多重要的数据,但并未纳入评估范围。这种评估方式的局限性是造成晚期失败的因素之一,而数字技术的应用可能会改变这一现状。
以心力衰竭[ 尤其是保留射血分数的心力衰竭(heart failurewith preserved ejection fraction,HFpEF)] 患者为例,这类患者虽然面临着巨大的疾病负担和生活质量的显著下降,但适用于监管部门审批的临床试验终点(例如死亡率)的发生率却非常低,尤其是在HFpEF 早期阶段,此时患者可能主要表现为器官功能受损。当研究依赖于传统的临床试验终点时,由于需要较大的样本量才可能观察到统计学上的显著性,导致针对此类患者群体的研究通常较难成功。因此,一些制药企业甚至放弃了对这类疾病的新药研发,从而阻碍了对此有需求的患者群体的医疗创新[4]。
1.2 DHT 驱动下的生物标志物与临床结局评估
基于现有临床试验终点的局限性, 药物创新需要变得更加聚焦于患者, 随着医药行业的数字化进程加速,医药研究、诊断和治疗的面貌正在经历根本性的变化。这一转型促进了数字健康技术(digital health technologies,DHT)的迅猛发展和广泛应用,更为患者、研究者以及医疗服务提供者提供了在传统临床环境之外收集健康相关数据的能力,且在收集衍生的数据时,患者的身体参数等数据具有作为替代终点支持药物临床研发的可能。
美国《处方药使用者付费法案》(Prescription Drug User Free Act,PDUFA) 的更新版PDUFA VI 要求美国食品药品监督管理局(Food and DrugAdministration,FDA) 将“ 患者体验”纳入新药的获益与风险评估,并成为推动药物研发不可或缺的重要部分。这一要求强调了了解和量化患者获益的重要性,同时PDUFA VI 也指出了需要利用新方法、新工具来衡量这些获益,包括可穿戴设备、医疗应用程序以及机器学习程序等[5]。
这些新方法、新工具不仅可以定量地捕捉患者体验,还能以定性的方式深入理解和分析患者的日常生活和治疗过程中的细微变化。对患者体验的全面捕捉和分析,能够在多个层面上转变医药产品的研发思维及过程,包括但不限于改进临床试验设计、提高治疗方案的个性化以及提升患者满意度等。通过这样的方式,医药行业能够更好地满足患者的具体需求,同时也符合监管部门“以患者为中心”理念的要求。
在药物研发过程中,有2 个主要的评估方法被用来提供支持药物批准的安全性和疗效数据,即生物标志物(biomarker) 和临床结局评估(clinical outcome assessment,COA)。生物标志物的概念于 1983 年首次提出,系指能客观测量并评价正常生物过程、病理过程或对药物干预反应的指示物。生物标志物是从客观评估中得出的测量结果,可以是体外(例如使用实验室仪器从生物样本中获得)或体内(例如通过成像技术或电生理常规技术获得)。1998 年,美国国立卫生研究院(National Institutes of Health,NIH)生物标志物定义工作组制定了生物标志物的定义,并确定了生物标志物的类别(例如诊断、预后或治疗反应),以及基于生物标志物的替代终点的定义。此后,生物标志物开始作为药物研发工具被广泛应用。2004年,FDA 认识到提高药物研发效率的重要性,并将生物标志物及相关的转化策略视为优先事项。目前,生物标志物的使用已成为药物研发的主流[6]。
COA 与生物标志物不同,其捕捉了治疗手段对患者感受、功能或生存的明确识别方面的影响数据。COA 根据报告者进行分类,通常患者报告结局(patien treported outcomes,PRO)、临床医生报告结局、表现结局或观察者评估结局的形式收集。COA 与生物标志物的主要区别在于,COA 捕捉了患者重要的病情以及影响患者生活方式等方面的数据,并可能受到个体的有意识选择、判断或动机的影响[7]。美国卫生当局提出的以患者为中心的药物研发概念增加了对患者和照顾者有意义的评估的重视。COA 和生物标志物是临床研究的重要组成部分,二者的有效性、可靠性和对变化的敏感性提示其可以成为药物临床研究的替代终点,还可以作为药物安全性和有效性的重要依据。
通常,FDA 批准的临床试验终点关注于患者的感受、功能状态或生存期,并且已经为药物研发过程中的生物标志物或 COA的资格认定和使用给出了非常明确的指导和途径,使其能够成为临床试验替代终点。DHT 在实际应用中能够产生丰富的数据,其衍生测量可以捕捉到多种关注概念, 反映多个关键的健康指标,如图1 所示。在某些情况下,DHT 衍生的数据构成了传统的生物标志物,例如通过心电图测量的心率或通过远程脉搏血氧仪测量的血氧饱和度等,这些数据的本质与生物标志物的定义相吻合。在其他情况下,DHT 衍生的数据可以构成COA,例如通过可穿戴设备收集的6 分钟步行试验(six minutes walk test,6MWT)等。DHT 衍生的数据最终被归类为生物标志物还是COA 取决于以下2 点:①数据是否作为某种疾病/ 病情的病理过程的指标,具有对干预或暴露响应的特征。②数据是否与患者的表现或功能相关联,是否具有对患者有意义的概念。数字生物标志物和数字COA可以成为替代临床试验终点,支持药物上市,例如步幅速度第95百分位数(stride velocity 95thcentile,SV95C)支持在杜氏肌营养不良症药物研发中的应用。
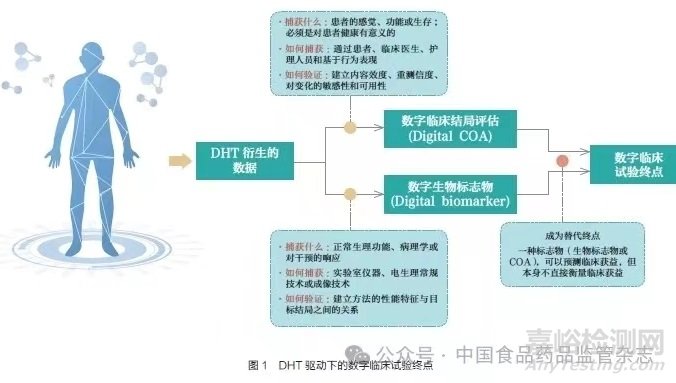
1.3 支持数字临床试验终点的相关技术
支持数字临床试验终点的技术类型可分为3 种,包括可穿戴设备(例如腕戴式加速度计、手指佩戴传感器和生物识别皮肤贴片等),智能手机或应用程序(例如语音分析和打字行为等),非可视技术(即无需患者佩戴任何传感器,可被动测量的技术)。非可视技术包括Sensor-enabled Homes 和EmeraldTM,其利用的是无线电波反射信号与机器学习算法,见表1。

2 药物临床研发中数字临床试验终点的应用
2.1 杜氏肌营养不良症
2019 年,SV95C 成为第一个满足监管要求的数字 COA,SV95C 已获得欧洲药品管理局(European Medicines Agency,EMA)认可,可用于杜氏肌营养不良症关键研究的次要终点,且还被纳入FDA 的 COA 资格计划中进行审查。例如ActiMyo® 是目前唯一一个经验证适用于在临床试验中捕捉 SV95C 的被动式设备, 应用ActiMyo® 的患者在日常生活中不需要完成任何任务。该设备已经在控制和非控制环境下,针对对照组和患者群体进行了广泛的验证。算法可将设备的数据转化为物理变量(例如步长和速度),再根据物理变量计算出临床变量(例如SV95C)。EMA的资质认证确认了SV95C 的准确性、可靠性、对变化的敏感性以及与患者的相关性。更具体地说,SV95C 能够连续地量化患者在家庭环境中的最大步行速度,并反映6MWT 的大部分组成数据(除了耐力等附加信息以及评估时的动机或疲劳等干扰因素之外)[8]。
ActiMyo® 可穿戴设备联合SV95C 数字终端, 共同构成了一个跨越医疗器械与药品监管框架的数字治疗技术工具(digital health technology tools,DHTT)。在研发阶段,研究人员遵循了包括欧盟医疗器械法规 (EU)2017/745 和(EU)2017/556,以及一系列国际相关标准。这款可穿戴传感器作为一种I 类医疗器械,符合欧盟CE 标志的要求,并在ISO13485:2016《医疗器械 质量管理体系 用于法规的要求》(Medical Devices-Quality Management Systems-Requirements for Regulatory Purposes)质量管理体系的指导下进行研发。
EMA 通过资格意见书认可了SV95C 作为适用于5 岁以上杜氏肌营养不良症患者的临床试验的次要终点[9],前提是使用了合适的可穿戴设备进行测量。这份资格意见书不仅涵盖了ActiMyo® ,还包括了其硬件和软件的性能。
这项资格申请是由一支跨学科的专家团队共同准备的, 包括儿科神经学家、生物统计学家、物理治疗师、工程师、法规事务专家以及患者代表。该团队于2017 年6 月提交了资格申请意向书,最终的资格意见在2019年4 月被EMA 的人用药品委员会(Committee for Medicinal Products for Human Use,CHMP)采纳。这次资格申请的依据来自于数个全球性的自然历史研究和关键试验。目前,该团队已向 EMA 提交了一份后续资格意见申请,以升级将 SV95C用作临床试验终点,并将 SV95C的应用推广到其他以近端肌无力导致步行困难逐渐加重的神经肌肉疾病。
2.2 多发性硬化症
多发性硬化症(multiple sclerosis,MS) 是一种影响中枢神经系统的病症,表现为炎症导致的神经纤维脱髓鞘以及随后的神经退行。MS 的发展模式因人而异,表现出广泛的临床差异[10]。通常, 对MS 的监测是间歇性的,缺乏一致性,主要依赖于临床测量, 例如扩展残疾状态量表(expanded disability status scale,EDSS) 和磁共振成像等。这些方法的局限性使得病情进展的早期识别成为挑战[11]。在MS 早期,诸如认知功能下降等方面的变化可能是微妙的,且属于亚临床水平,但随时间推移,其发生频率和严重程度可能会增加。疾病恶化是一个复杂的多维过程,较难准确捕捉。目前,MS进展的诊断大多是回顾性的,主要依赖患者的临床病史,并要求EDSS 评分显示至少6 个月的连续恶化,且无复发迹象。MS 的监测通常依赖于不频繁的门诊评估(一般每年1~2 次),且医务人员在客观评估病情进展方面的工具有限[12]。因此, 迫切需要新的临床和研究工具,以实现对MS 早期病变和病情持续恶化的有效监测,这也为数字健康领域创新带来了机会。
为了改善MS 的疾病进展测量,2 家生物技术公司达成了一项合作协议,旨在通过2 款基于智能手机的应用程序提升数字临床试验终点的监管认可机会。这2 款应用程序运用了相似的概念,包括一系列的主动和被动测试,例如认知测试、形状绘制、捏合力测试、两分钟步行和U 形转弯测试等,用以全面评估患者在认知、上肢功能、步态、平衡和总体移动性等方面的能力[13]。其中,Floodlight™ MS 由包括智能手机应用在内的数个具有欧盟CE标志的医疗设备软件(medical device software,MDSW) 组成, 这些设备旨在客观地监测MS 患者在2 次临床访问间的功能表现;Konectom™也是一款基于智能手机、获得欧盟CE 标志的MDSW,其提供9 项评估功能,旨在通过患者自述和性能评估相结合的方式,准确量化MS 患者的神经损伤,包括运动和认知功能损害[14]。
Floodlight™ MS 和Konectom™应用程序均可作为数据收集工具,在临床试验中用于评估治疗效果,并在日常临床实践中辅助患者管理,为医务人员提供关键的护理信息。这种并行的研发策略意味着测量工具不仅可在药物研发阶段使用,还可以在日常治疗中应用,这样可以收集到更高质量的真实世界数据,为患者提供更及时的治疗选择。
Floodlight™ MS 和Konectom™应用程序在欧盟受医疗器械法规(EU)2017/745 的管理, 并被归类为Ⅱ a 类MDSW。当这些应用程序用于药物临床试验时,其需要遵守良好临床实践(good clinical practice,GCP) 标准,尤其是计算机软件验证[15] 的要求。从这些智能手机应用程序中得到的数字临床试验终点可能需要经过资格认定程序。为了探索上述可能性, 应用程序开发商与EMA 举行了创新任务组(Innovation Task Force)会议,针对Floodlight™ MS 和Konectom™衍生的数字临床试验终点是否有资格成为临床终点进行讨论。
2.3 特应性皮炎
瘙痒(痒)是许多慢性湿疹病症的主要症状,尤其在特应性皮炎(atopic dermatitis,AD)患者中更为常见。对瘙痒感觉的常见反应是搔抓受影响的区域,这会导致额外的炎症或损伤形成,从而加重瘙痒并延续痒- 抓循环。此外,瘙痒通常发生在晚上和夜间,会严重影响患者睡眠。痒-抓循环加上睡眠障碍更降低了患者和照顾者的生活质量。瘙痒和睡眠的传统评估主要基于COA和PRO 指标。COA 旨在评估病变的总体表面积以及病变的严重程度(红斑、硬结、擦痕等),但这些指标主要由医务人员衡量,对于患者在诊所外经历的症状波动仅能提供有限的见解。相比之下,虽然 PRO 提供了患者角度的感知状况,但其是主观的,可能受情绪或建议的影响,缺乏遵从性,并且具有定性的特点。因此,需要更多客观的测量指标,准确反映 AD 对患者日常生活的影响。这些客观的测量指标不仅有潜力提供更可靠的干预效果指标,还可以帮助改善疾病的管理。
为了客观测量患者夜间搔抓行为和睡眠的情况,研究人员开发了一种手腕佩戴式可穿戴设备。这种设备能够通过加速度计数据来监测和记录患者的夜间搔抓行为。研究人员采用启发式和机器学习算法对加速度计数据进行处理和分析,以识别睡眠和搔抓事件,并以睡眠实验室收集的数据作为参考标准,对算法进行了验证。共有45 名AD 患者被招募参与研究,其中33 名AD 患者的数据被用于最终分析。这些AD患者在睡眠实验室中接受了监测,并在家中使用了手腕佩戴式设备。结果显示,通过手腕佩戴式设备收集的数据在测量总睡眠机会和总睡眠时间方面与多导睡眠图数据高度相关。此外,该设备记录的搔抓持续时间与视频记录的标注数据也具有较好的一致性[16]。
2.4 癌性恶病质
癌性恶病质是一种常见的代谢紊乱,其影响着大多数晚期癌症患者,主要表现为肌肉流失、体重下降以及持续疲劳等症状。这种症状群不仅对患者的生存构成威胁,还严重影响其生活质量,尤其是在身体功能方面。尽管癌性恶病质对患者的生活带来了重大挑战,但目前尚没有针对性的批准药物[17]。根据现有的定性研究证据,癌性恶病质患者迫切希望能够提升自己的身体功能,特别是独立进行锻炼和日常活动的能力[18]。然而,目前的临床试验未将相关终点纳入研究,这表明在癌性恶病质的临床研究中,迫切需要有效的评估工具来准确衡量身体功能的变化。
现有的评估方法存在局限性,其通常只能间歇性地评估身体功能,给患者带来额外负担,并且可能缺乏现实生活环境中的有效性[19],DHT 能够克服传统评估方法的不足,提供更加全面和连续的监测手段。作为DHT的一种,可穿戴传感器在伴随肿瘤出现的消瘦症状研究中发挥着重要作用,其能够实时追踪患者的身体活动。过去, 研究人员主要依赖于临床测试来观察肌肉质量减少与身体功能下降之间的关系。现在,可穿戴传感器的使用使得研究者能够客观地、持续地监测癌性恶病质患者的身体行为模式。这些传感器能够详细记录患者在各种日常活动中的时间分配,例如坐立、躺卧、站立、行走以及从坐到站的转换。activPAL 是一种佩戴于大腿的加速度计设备,其已经在癌症患者中验证了可行性和基于标准的有效性。activPAL 能够精确测量不同功能水平(例如卡氏状态评分)下的身体姿势和运动,为评估癌性恶病质患者的活动能力和生活质量提供了新视角。通过这些技术,研究人员能够更准确地理解和评估癌性恶病质患者的日常活动模式,从而为治疗和护理提供更有针对性的支持[20]。
3 总结与展望
DHT 在药物研发领域的重要潜力正在逐渐展现。这种技术能够让药物研发人员获取连续且客观的数据集,特别是在临床环境之外,以及那些能够更真实反映患者日常生活的活动,这是传统技术难以实现的。通过采用数字临床试验终点, 研究人员能够更早地获取敏感和可靠的安全性及疗效指标, 有助于精简和缩短临床试验进程, 以及加速决策过程。
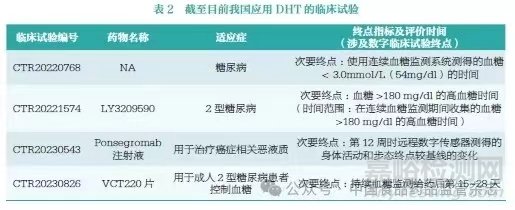
在药物研发后期,数字临床试验终点不仅对于内部决策具有重要意义,而且有潜力成为药物注册申请中的主要或次要终点,甚至帮助支持药物说明书。尽管存在较多优势,数字临床试验终点在临床试验中的广泛应用仍面临着一些挑战和障碍。大多数应用案例仍处于试点阶段或作为探索性临床试验终点进行研究。尽管如此,已有部分案例研究结果表明,数字临床试验终点作为后期临床试验的主要或辅助终点开始被接受和应用。截至目前,我国有4 项临床试验中使用过数字临床试验终点,见表2。整体来看,我国使用DHT 的临床试验相对较少,尚未有基于数字临床试验终点获批的药物,我国数字临床试验终点尚处于发展初级阶段。为此,本文提出了几点建议,以期能够加速我国DHT 在临床试验中的应用与发展。
3.1 加强数字临床试验终点监管科学研究
在规划临床试验时,由于缺乏关于DHT 衍生终点的指导原则,将数字临床试验终点纳入关键试验终点存在一定挑战。尽管2022 年国家药品监督管理局药品审评中心发布了《患者报告结局在药物临床研发中应用的指导原则(试行)》,为行业开发新终点提供了方法与路径。但仍缺乏基于数字临床试验终点的具体指导原则,申办方较难确定获得监管批准所需的证据量,这使得数字临床试验终点的整体规划变得不确定,降低了行业对数字临床试验终点应用的积极性。同时,当前企业基于DHT 的创新和终点研发速度可能已经超过了相关法规要求,监管部门需要加强关于数字临床试验终点的监管科学研究,加速制定相关标准,发布数字临床试验终点的指导原则,促进行业研发可有效反映患者疾病负担的数字临床试验终点。
3.2 建立数字临床试验终点认证程序
根据美国《21 世纪治愈法案》(21st Century Cures Act), 在《联邦食品药品和化妆品法案》(Federal Food, Drug and Cosmetic Act)中增加了“药物开发工具资格认定程序”条款,使原来FDA以指南形式制定的DHT 具有法律效力。在DHT 中,具有代表性的工作成果是COA 认证和生物标志物资格认定计划[21]。通常,FDA 批准的临床试验终点关注于患者的感受、功能状态或生存期,经过认证的生物标志物和COA可以成为临床试验替代终点,直接在临床试验中使用,很大程度上降低了企业的研发成本。
目前,我国尚未建立当前生物标志物和COA 的资质认证相关程序,而生物标志物和COA的研发以获得监管部门的认可是一件耗时和繁琐的工作,制药企业更愿意将资源集中在临床试验数据的生成与收集方面,这在很大程度上阻碍了临床试验替代终点的研究。因此,建议我国基于国内数字化进程,合理建立数字临床试验终点认证程序,促进行业开发使用数字临床试验终点,使临床试验更加以患者为中心。
第一作者简介
张新宇,硕士,荣昌生物制药(烟台)股份有限公司,注册主管。专业方向:药品监管科学
通讯作者简介
程龙,博士,荣昌生物制药(烟台)股份有限公司,副总裁。专业方向:创新药研发,临床研究与注册,监管科学研究
参考文献:略

来源:中国食品药品监管


