您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-08-18 11:26
摘 要 / Abstract
改良型新药在美国按505(b)(2) 路径申报和审评,相关药物的销售市场规模约为仿制药的1.7倍,在满足临床需求的前提下,505(b)(2) 药物因竞争压力较小而具有较大的经济效益。近年来,我国仿制药市场增长较为缓慢,越来越多的仿制药企业希望通过布局改良型新药获得新的增长点。同时,随着创新药获批数量的快速增长,越来越多的创新药企业开始利用改良型新药的路径增加产品的适用人群或延长生命周期。然而,改良型新药是一个具有挑战性的全新领域,不仅审批标准较高,而且产品设计的理念和经验尚不成熟,导致相关项目开发失败率较高或市场收益不理想。美国505(b)(2) 路径已发展近40 年,覆盖2500 多个批准文号,且很多产品的市场开拓取得成功,因此美国505(b)(2) 路径的设计与开发、药物临床评价及市场开拓对我国具有一定的借鉴意义。本文系统对比了我国改良型新药与美国505(b)(2) 路径药品申请的异同,梳理了美国505(b)(2) 路径审评审批情况,分析了成功通过505(b)(2)路径上市的产品所具备的共性及其设计思路与亮点,以期为我国改良型新药的业务布局、项目设计和产品开发提供一定的参考。
Improved new drugs are applied and evaluated according to the US 505(b)(2) pathway, and the market of 505(b)(2) products is about 1.7 times that of generic drugs in the USA. While meeting clinical needs, the 505(b)(2) products present significant economic benefits due to less competition. In recent years, due to the slow growth of the Chinese generic drug market, more and more generic pharmaceutical companies are looking to improved new drugs for new growth points. In addition, with the rapid growth of innovative drugs, many innovative pharmaceutical companies are starting to apply this pathway to increase the potential patient population or extend the product lifecycle. However, developing improved new drug is a challenging field for Chinese pharmaceutical companies, with high approval standards and immature product design concepts and experience, resulting in a high failure rate of development or unsatisfactory market returns. In contrast, the US505(b)(2) has a forty-year history, over 2500 approvals, and many products have obtained successful market performance. Therefore, the design and development, clinical evaluation, and market expansion of the US 505(b)(2) pathway are valuable for reference. This article systematically compares the similarities and differences between China’s improved new drugs and the US 505(b)(2) pathway, reviews 505(b)(2) approvals, including their market performance, design ideas, and highlights, to provide references for the industrial planning, project design, and product development of improved new drugs in China.
关 键 词 / Key words
505(b)(2) ;改良型新药;设计;开发;市场
505(b)(2); improved new drugs; design; development; market
1、美国505(b)(2) 的批准文号统计和市场概况
1984 年9 月,美国通过了《药品价格竞争与专利期补偿法案》(Drug Price Competition and Patent Restoration Act),又称为Hatch-Waxman 法案,修订后的《联邦食品药品和化妆品法案》(Federal Food, Drug and Cosmetic Act,FDCA)505 部分为药物上市申请提供了3 条路径:505(b)(1)、505(b)(2) 和505(j)。其中,505(b)(2) 旨在为主活性成分与市售药品相同,但剂型、规格和配方工艺等方面与市售药品存在差异,不能归类为仿制药的产品提供一种相对简化的新药申请通道[1-2]。按目前美国食品药品监督管理局(Food and Drug Administration,FDA) 的分类, 化学药品的新药上市许可申请(new drug application,NDA)可分为10 种类型,除了1 类属于505(b)(1) 路径之外,2~10 类均属于505(b)(2) 路径(表1)[3]。
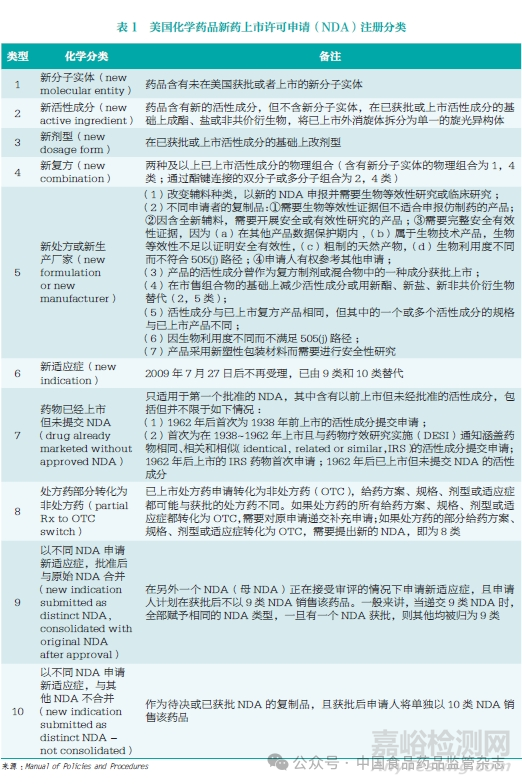
为了便于研究和分析,本文统计了近40 年以来美国FDA 批准的新药批文情况。统计结果显示,FDA 在1984~2023年仅批准了1328个新分子实体或新生物制品,低于授予的505(b)(2) 文号2523个。其中,2类90个,3类1252个,4类293个,5类705个,6类、9类、10类共112个,7类39个,8类32个。从趋势上来看,FDA每年批准的505(b)(2)数量相对稳定,有轻微的增长趋势,平均每年为63个,最多的一年为2016年的89个,最少的一年为1991年的29个(图1)。
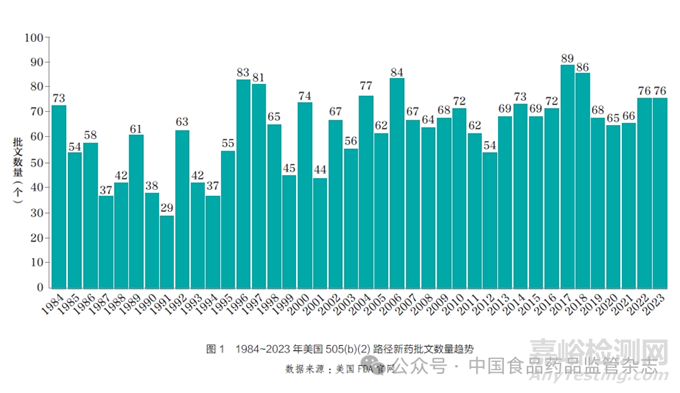
为了解通过505(b)(2) 路径上市产品的市场情况,本文使用艾昆纬(IQVIA)的全球药品销售数据库(Midas) 对1984~2023 年获批的2523个505(b)(2)产品批文进行一一检索,结果显示美国505(b)(2) 产品在2022 年的总销售额约为1050 亿美元(截至本文统计时,数据库尚未更新至2023 年,故尚无2022年下半年和2023 年批准的部分产品数据),约占美国医药市场的17%。从产品获批上市的时间来看,市场主流产品是在2010~2019 年获批的,销售额占59% ;其次是在2000~2010 年获批的产品, 销售额占29%。2022 年,2~5类505(b)(2) 产品的总销售额为924 亿美元;2017~2022 年, 复合增长率为1.04%,市场处于缓慢增长状态(图2)。对于6~10 类产品,由于利用数据库无法有效区分销售额或涉及大量院外非处方药(OTC)产品而参考价值有限,本文不做重点探讨。
需要注意的是, 在上述2523个505(b)(2) 批文中, 有702个在2022 年的销售额超过100 万美元, 占比不足30%。也就是说, 在1984~2023 年获批的505(b)(2) 批文中, 超70% 在2022 年没有销售额或销售额不足100 万美元。其原因一方面在于时间跨度太长,大量产品已经退市、不再销售或因专利问题无法上市。FDA 橙皮书数据库显示,在2523个批文中,36% 处于停止销售状态,4% 处于暂时性批准状态。另一方面,505(b)(2) 产品需要企业从头开始进行市场推广,只有少数品种能够脱颖而出。
在数据可区分度较高的2~5 类505(b)(2) 产品中,2022 年美国市场销售额排名前50 的产品销售额之和占总销售额的72.34%,市场集中度较高。在这50个产品中,36个被药物分子的原研厂家持有;9个被50强制药企业但非药物分子的原研厂家持有;仅5个被一般药物递送技术公司或制剂技术公司持有,销售额之和仅占上述50个产品的4.53%。销售排名在50 名后的产品市场格局较为分散,平均销售额仅为1100 万美元。
2、成功的505(b)(2) 产品共性分析
如上文所述, 虽然美国批准的505(b)(2) 产品数量较多,但年销售额超1 亿美元、生命周期内的总销售额超10亿美元的重磅产品屈指可数,且这些重磅产品大多来自于药物分子的原研厂家或制药巨头。造成这种结果的原因有两个方面:一是原研厂家拥有得天独厚的优势;二是美国505(b)(2) 产品的市场开拓投资巨大,对中小企业而言是一种挑战。
首先,原研厂家开发505(b)(2) 产品是为了品牌延伸,以增加适用人群或提升用药便捷性为目的。例如,强生在利培酮片上市之后,又开发其口服液和口崩片,其临床优势是建立在新分子实体之上的,更容易获得市场认可。其次,原研厂家开发505(b)(2) 产品是为了延长产品的生命周期,可在药物化合物专利到期前停止推广普通制剂,同时切换为升级产品,如阿斯利康在奥美拉唑专利到期前,将奥美拉唑的处方替换为埃索美拉唑。再次,原研厂家或制药巨头拥有强大的品牌效应和渠道资源,学术推广能力及推广专业度也非一般药物递送技术公司或制剂技术公司所能及。最后,原研厂家拥有全面的药动学、药效学数据和用户反馈,能够直击患者痛点,找到最优的改良方案。相反,一般公司的505(b)(2) 产品往往面临诸多挑战。一是品牌要从头开始推广,若无足够临床优势和创意,就很难获得医生的关注和认可。由于大部分药物递送技术公司或制剂技术公司是中小型企业,渠道能力和推广能力都远不如原研厂家或制药巨头,很难做大市场。二是如果没有足够的临床额外获益,高价的505(b)(2) 产品会受到药品福利管理(PBM)组织的抵制,进而很难被纳入医保报销范围。三是505(b)(2)产品必须具有不可替代性(产品与仿制药存在显著差异或医生开具处方时注明不允许替代)才能产生销量,否则会在配方时被药师用仿制药替代。
鉴于原研厂家的优势和中小型药物递送技术公司或制剂技术公司的劣势,后者通常会积极与原研厂家合作,为原研厂家的产品升级提供方案,或与在项目所属领域拥有强大管线的制药巨头合作,把产品委托给制药巨头销售。然而一般情况下,制药巨头仅对有技术创新、专利保护和临床价值的改良型新药接受销售委托,并投入巨资开拓市场。因此,如果产品没有显著优势,将可能无法产生销售额。
虽然一般药物递送技术公司或制剂技术公司持有的产品很难发展成为重磅产品,但其中依然有成功突围的产品。基于对这些产品的梳理与分析,可以归纳出以下几点共性:富有创意的设计,技术含量较高或能形成强有力的专利保护,巨大的临床优势或能够解决临床痛点,有足够的市场卖点。
2.1 富有创意的设计
例如, 环孢素滴眼液(Restasis)是一款畅销的505(b)(2) 产品,其创意来自动物实验以及发现肾移植患者用药后泪液明显增加,后来进一步研究发现炎症能够抑制泪液释放,而环孢素作为免疫抑制剂,可以调节免疫系统而干预炎症。再如,右美沙芬可调节N- 甲基-D- 天冬氨酸(NMDA)受体,但容易被肝药酶代谢,Avanir 公司将其与肝药酶抑制剂奎尼丁联用并开发了Nuedexta,获得FDA 批准用于治疗假性延髓情绪。多年后,该创意被二次利用,Axsome 公司将右美沙芬与肝药酶抑制剂安非他酮联用并开发了Auvelity,成为治疗成人重度抑郁症的突破性疗法。此外,纳洛酮是一种阿片受体阻断剂,因没有选择性而无法用于阿片类药物引发的便秘治疗(需选择性阻断外周阿片受体),但经过聚乙二醇(PEG)修饰后便无法进入中枢神经系统,不但有效解决了阿片类药物引起的便秘,而且不影响阿片类药物的治疗效果。
如果设计者无法跳出“仿制”的思维局限,就难以开发出富有创意的产品。由技术简单类推的创意(如片剂改缓释片),除非技术壁垒极高,否则很容易与他人“撞车”或被他人复制,因为这种“创意”也能被其他技术人员想到和做到。创意通常来自生活,设计者需走出实验室,与医生、患者和销售人员交流他们的痛点,从中发掘设计灵感。
2.2 技术含量较高或能形成强有力的专利保护
一般情况下, 产品的复杂程度越高,生命周期就越长,因为这些产品很难被仿制,如亮丙瑞林微球、奥曲肽微球等。然而这样的产品非常罕见,绝大部分505(b)(2) 产品都躲不过被仿制的命运,因此强有力的专利保护是产品成功的关键。有研究者统计分析了2010~2020 年美国批准上市的3~5 类505(b)(2) 产品获专利保护的占比:3 类产品为85.7%,4 类为77.4%,5 类为50.3%[4]。这表明随着技术含量的下降,获专利保护的概率也在减小。在不同类型505(b)(2) 产品中,5 类的开发门槛最低,获得专利保护的概率也最小。
事实上,对于简单改剂型、配方或工艺的产品,即便获得了专利授权,保护效力也并不理想。因为仿制药厂家即便不使用专利保护的配方和工艺,也可能实现生物等效,所以技术门槛不够高的口服505(b)(2) 制剂,即便获得了市场认可,大多也只是昙花一现。除了专利保护,数据保护也是阻止仿制药进入市场的有效手段。根据美国的相关法规,只有开展了临床试验(不含生物等效性试验)的NDA才能获得3 年的数据保护期,但很多“改动不大”的产品容易被豁免临床试验,获得数据保护期的概率也很小。有研究统计分析数据显示,3 类505(b)(2) 产品获得数据保护期的比例为57.1%,4 类为66.0%,5 类仅为32.3%[4]。
如果既没有强有力的专利保护、数据保护,也没有足够高的技术壁垒,产品即便获得了市场认可,也会很快迎来仿制药的激烈竞争。因此,5 类505(b)(2) 产品虽然研发难度较小,开发和注册成本也相对较低,但其生命周期通常比一般产品更短,市场影响力也更小,平均销售额仅为0.24 亿美元,低于2~5 类产品的平均水平(0.37 亿美元)。
2.3 巨大的临床优势或能够解决临床痛点
要想让产品拥有足够的临床优势,在产品设计之时,就必须以获得临床优势为出发点。例如,降低药品的不良反应(如脂质体阿霉素相比普通注射剂降低了心脏毒性风险,白蛋白结合型紫杉醇相比普通注射剂杜绝了对紫杉醇发生超敏反应);提高患者依从性(如奥氮平口崩片相比普通片剂克服了吞咽障碍,双氯芬酸钠喷雾剂相比口服制剂使用更便捷);增加用药便捷性,扩大用药场景(如预充式肾上腺素注射液可以作为患者家庭和公共场所的储备用药,每日一片药物的艾滋病“鸡尾酒疗法”可以大幅减少患者服药错乱的现象);提高疗效(如丁丙诺啡缓释注射剂相比口服疗法可显著提高毒瘾戒除成功率,白蛋白结合型紫杉醇可以提升乳腺癌治疗应答率);二次定位产品,填补用药空缺(如芬太尼透皮贴剂可用于癌痛管理,硝酸甘油凝胶剂可用于局部治疗勃起功能障碍);防止阿片类药物滥用(如防滥用制剂羟考酮);大幅降低治疗成本(如口服抗生素可用于序贯治疗,减少患者的住院时间)。
总之,为了体现产品的临床优势,需要通过临床试验来证明,不能吝啬必要的临床投入。若想节约研发成本,在产品设计之时,不仅要在药物机制上解释清楚潜在的临床优势,还需要有可以量化的指标。例如,虽然非诺贝特通过增溶技术实现了给药剂量下调,提高了生物利用度,但并没有直接、可量化的指标证明其临床优势。
2.4 有足够的市场卖点
影响产品卖点的因素很多,如是否会为患者带来新的痛点,是否可稳定、大批量生产,用药成本是否过高,是否会引起严重不良反应,是否符合患者的生理特征和使用习惯,是否相较于竞品有独特的优势,产品创意是否为消费者所需要等。例如,基因泰克和Alkermes 公司联合开发的生长激素长效微球,虽然可以大幅减少患者打针的痛苦,但因为生产成本过高,上市后就很快退市。再如,可监视药物体内处置过程的创新制剂——带传感器的阿立哌唑(Abilify MyCite)几乎没有产生销量,因为这种创意对于大部分消费者而言是华而不实的。
3、505(b)(2) 路径的审评审批情况分析
505(b)(2) 产品或改良型新药是基于已上市活性成分的二次开发,大部分安全有效性证据是已知的,故不需要开展完整的安全有效性试验。由于不同国家和地区的药品审批标准不同,申请人所需开展的试验项目也有所差异。在505(b)(2) 路径的申报过程中,申请人可以通过新药临床试验申请前(pre-investigational new drug,Pre-IND)会议与美国FDA 建立有效的沟通以尽量豁免或减免临床试验,甚至豁免生物等效性试验,从而大幅压缩开发时间和成本[5]。有研究者统计分析了2010~2012 年美国FDA 批准的106 项不涉及美国总统防治艾滋病紧急救援计划(U.S. President's Emergency Plan for AIDS Relief,PEPFAR)的505(b)(2) 申请,其中有26 项(25%)仅基于临床药理学数据就获得了批准,有29 项(27%)被豁免了生物等效性或生物利用度试验和临床药理学试验,仅43% 的申请被要求开展临床药理学试验和临床试验[6]。
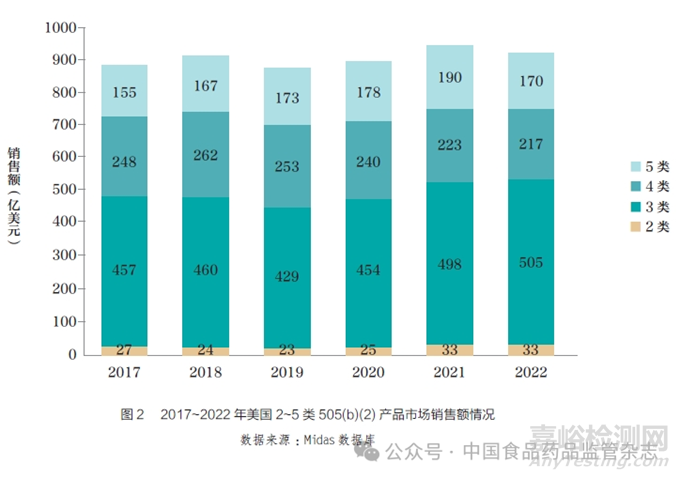
从剂型角度来看,有研究者统计分析发现,2012~2016 年获批的226 项505(b)(2) 申请中,拥有完整审评信息的3~5 类产品申请有112 项,其中口服速释制剂、口服缓释制剂、口服迟释制剂(肠溶制剂)、口服新复方制剂、注射剂、变更辅料、新增规格和改变给药途径的申请分别为20 项、18 项、5 项、18 项、9 项、20 项、11 项和11 项。结果显示,上述申请中,获得生物等效性或生物利用度试验豁免的分别占比20%、0%、0%、0%、33%、75%、9% 和18% ;需要开展安全有效性研究(不含特定安全性研究)的分别占比25%、61%、80%、94%、0%、10%、45% 和36% ;需要开展临床前研究的分别占比5%、11%、20%、33%、20%、50%、27% 和55%[7]。从这些数据不难看出,美国FDA 对口服速释制剂、注射剂、变更辅料的505(b)(2) 申请,要求相对较低(图3)。
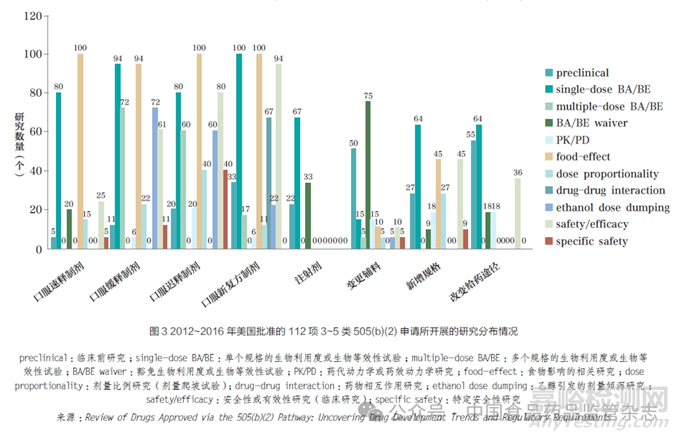
审评速度方面,有研究者统计分析了284个在2009~2015 年获批的505(b)(2)产品,平均审评时间为18.4个月。相较于创新药,505(b)(2) 产品在第一轮审评结束后获批的概率较低,仅为49%。其中,有31 项获得了优先审评资格,9 项获得了罕见病用药认定[8]。
4、我国改良型新药与美国505(b)(2) 产品异同
2015 年8 月,国务院印发《关于改革药品医疗器械审评审批制度的意见》[9],原国家食品药品监督管理总局根据相关要求于2016 年3 月发布《化学药品注册分类改革工作方案》[10],我国对改良型新药首次有了明确的规定。我国改良型新药虽与美国505(b)(2) 产品有诸多相似之处,但其中注册分类为2 类的改良型新药更强调临床优势。具体而言,美国505(b)(2)产品的临床优势主要由市场评判,我国2类改良型新药如没有显著性临床优势,就可能无法获得批准,甚至新药申请也可能不被受理。
我国2 类改良型新药主要分为以下几种:2.1 类为新活性成分,包括光学异构体,新盐、新酯,新酸根、新碱基或新螯合物,类似于美国505(b)(2) 产品注册中的2 类;2.2 类为新剂型、新处方工艺、新给药途径,类似于美国505(b)(2)产品注册中的3 类和5 类;2.3 类为新复方制剂,类似于美国505(b)(2) 产品注册中的4 类;2.4 类为新适应症,类似于美国505(b)(2) 产品注册中的6 类、9 类和10 类。
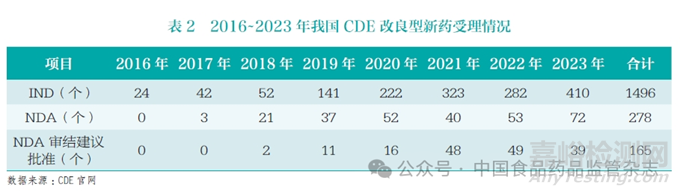
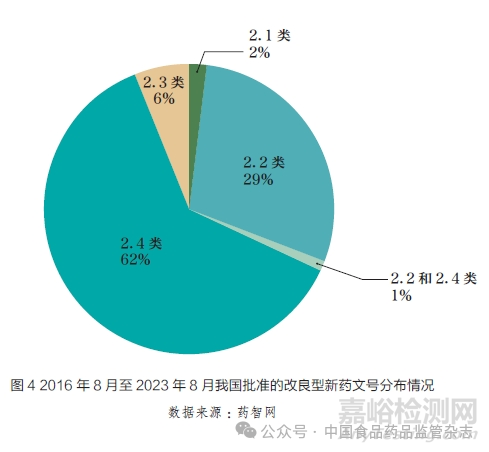
我国对2 类改良型新药的临床优势有明确的要求,相关注册申请通过率较低。本文基于国家药品监督管理局药品审评中心(Center for Drug Evaluation,CDE)历年发布的药品审评报告数据进行了统计分析,2016~2023 年,CDE共受理了1496个改良型新药的IND,仅有278个进入NDA 阶段,其中有165个审结并建议批准(表2)。另基于药智网数据分析发现,2016 年6 月至2023年8月,共有148个改良型新药文号获批,其中2.1类3个,2.2类42个,2.3类9个,2.4类92个,2.2和2.4类2个,分别约占2%、29%、6%、62% 和1%(图4)。与美国505(b)(2) 产品相比,我国改剂型、改配方的产品比重较低,新适应症产品比重较高。
除以上差异外,美国还对开展了临床试验(不含生物等效性试验)的产品提供3 年数据保护期,如果获得特殊认定则该期限更长,如罕见病用药认定的数据保护期为7 年,合格的传染病用药为5 年,儿科用药可额外增加0.5 年[5]。
5、我国改良型新药的市场状况
本文通过Midas 数据库对我国已批准的2.1~2.3 类改良型新药进行一一检索,结果显示共有22个品种在该数据库中拥有销售额,2022 年和2023 年上半年的总销售额分别为4830 万美元和4290 万美元。由于数据库无法区分适应症,在商品名、剂型、规格一致的情况下,无法判断销售额来自新药还是改良型新药,故与美国的6 类、9 类和10 类505(b)(2) 产品一样,也不将此类产品纳入研究。
尽管可获得的销售数据不多,但已有数据明显表现出增长趋势。由于我国改良型新药审批通道发展较晚,现有销售数据对市场状况的反映程度有限,本文不再做过多赘述。在不同类型的改良型新药中,2.1~2.3 类产品占比不足40%,这一方面说明我国药品监管部门对新剂型、新复方的审批标准较高;另一方面显示出,我国制药企业在载药技术和产品设计经验方面,相比美国505(b)(2) 产品申请人还存在一定的差距,相关项目开发成功率有待提高。
总之,越来越多的国内企业开始重视改良型新药的布局,有的创新药企业已经把这种路径作为提升受众基数、扩大产品销量、延长产品生命周期的有效工具。例如,先声药业在依达拉奉右莰醇注射用浓溶液获批后又开发了其舌下片;信立泰药业在面临阿利沙坦酯“专利悬崖”之际,又布局了多个新复方。而仿制药企业鉴于激烈的市场竞争,普遍将开发改良型新药作为一种出路。上海谊众的注射用紫杉醇聚合物胶束于2021 年获批上市,2023年的营业收入就达到了3.6 亿元,这大大增强了改良型新药对企业的吸引力。由于很多企业习惯开发仿制药,在改良型新药的设计方面,“仿”的元素明显大于“创”,从而引发多个赛道异常拥挤或出现“为改而改”“乱改剂型”等现象,这些都是改良型新药开发失败率较高、市场表现不理想的主要原因。
6、讨 论
虽然我国的改良型新药与美国的505(b)(2) 产品的审评和审批要求有所不同,但市场要求是一样的。因此,成功的505(b)(2) 案例可以为我国改良型新药的设计提供一定的参考。如果企业不是药物分子的原研厂家,在产品布局时不考虑成功505(b)(2) 产品所具备的共性(富有创意的设计、技术含量较高或能形成强有力的专利保护、巨大的临床优势或能够解决临床痛点、有足够的市场卖点),仅是为开拓管线而盲目撒网,那么成功的概率注定大打折扣。
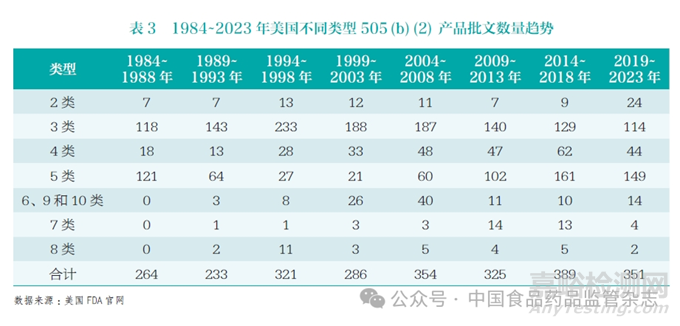
改良型创新不局限于改剂型,因为从载药技术上发掘临床优势的难度越来越大,美国3 类505(b)(2) 产品的申报活跃度已呈现出明显的下降趋势(表3)。首先,随着药物筛选技术的逐渐提高,药物分子原研厂家在分子设计上侧重于一步到位,留下的改良机会并不多。其次,由于创新药研发成本攀升,原研厂家非常重视产品生命周期的延长,便不断升级配方,使得改良的机会大幅减少。再次,载药技术鲜有重大突破,老药经过多次改良已再难有提升空间,而近年来新上市的产品可供改良的机会不多,因此产品设计方面出现了瓶颈。此外,常见载药技术已普遍被仿制药企业掌握,大部分505(b)(2) 产品因仿制药的快速出现而投资回报不佳。最后,鉴于美国的“阿片类药物保卫战”[11],FDA 收紧了对阿片类药物的监管[12-14],导致缓控释制剂的开发积极性迅速下滑。
改良型创新也不等于比拼技术难度。在美国,技术壁垒较低的5 类505(b)(2)产品的申报活跃度正在快速上升。从逻辑上讲,企业申报的5 类505(b)(2) 产品主要有以下几种类型。一是原研厂家为延伸品牌树和延长产品生命周期而申报大量的简单制剂,如强生在利培酮片上市后又推出了口崩片,艾尔建在贝美前列素滴眼液面临“专利悬崖”时升级了配方。二是因原研产品的配方专利保护或使用了特殊工艺而无法做到生物等效或处方组成/ 用量(Q1/Q2)一致,仿制药企业只改变策略选择申报5 类505(b)(2),如阿特维斯、费森尤斯和山德士等公司为了绕开卡巴他赛的配方专利而相继提交了NDA。三是在消费升级的趋势下,许多企业为满足高端用户的需求而升级配方,如把普通注射剂升级为预充式注射剂,把大输液升级为固液双室袋,将滴眼液升级为无防腐剂配方等。四是大输液厂家为丰富产品管线而开发大量新配方,如费森尤斯使用不同的油相配比开发了多个脂肪乳配方。
虽然5 类505(b)(2) 产品的研发门槛较低,但这并不代表其没有存在的价值。首先,企业以提升便捷性、依从性为目的,开发口服液、口崩片、咀嚼片以及小规格药品等,可有效解决老、幼以及肝肾功能不全等患者的用药痛点。其次,不同的患者对药物有不同的诉求,其中便捷性是一个重要的考量因素。例如,艾滋病治疗用药逐渐从一日多次、多片向一日一片发展,慢性病用药剂型向服用时不需要喝水或只需要少量水的口崩片、口溶膜发展等。最后,因原研产品存在组合物、晶型或工艺专利而不能申报仿制,仿制企业通过适度改工艺或配方申报505(b)(2),有利于促进竞争,进而降低药价,减轻患者经济负担。总之,或许这些产品的临床优势并不明显或难以量化,但其的确是市场所需要的。
我国改良型新药和美国505(b)(2) 产品都兼具“复制”和“创新”的属性[15],虽然在审评、审批上存在诸多差异,但我国的改良型新药也必将成为创新药和仿制药之间的一块独特而不可或缺的市场。对于普通药品研发企业而言,在仿制药受到药品集中带量采购等影响的情况下,改良型创新是一条既符合实情又有一定前景的转型路径;对于创新药企业而言,改良型创新是延长产品生命周期、扩大细分市场、增加受众人群的有效途径。
引用本文
魏利军,雷继峰*.美国505(b)(2) 路径对我国改良型新药设计与开发的启示[J].中国食品药品监管,2024(7):56-67.

来源:中国食品药品监管杂志


