您当前的位置:检测资讯 > 实验管理
嘉峪检测网 2024-08-26 13:15
本文对GC峰拖尾问题的成因进行分析并提出解决思路。峰拖尾是毛细管气相色谱法中经常遇到的问题,它可能导致分离度和峰积分问题,影响定性和定量分析。
以下就从三个步骤进行逐一阐述:
第一步判断峰拖尾的类型
气相色谱峰拖尾有许多种类型,但最主要的类型总结为以下四类:
第一类:所有峰都拖尾(包括溶剂峰)。
第二类:仅部分待测物质峰拖尾。
第三类:仅溶剂峰和(或)保留时间较短的峰拖尾。
第四类:仅保留时间较晚的待测物质峰拖尾。
以上每种情况下,色谱峰拖尾的严重程度可能从几乎无法辨别的拖尾(放大色谱量程可见)至严重拖尾(全量程时即明显可见),严重拖尾一方面影响检测灵敏度,另一方面影响相邻峰的分离度。拖尾的严重程度对问题来源将有重要的指示作用。
第二步识别峰拖尾的情形
第一类:色谱图中所有峰均拖尾。该种情况可能是最常见和最容易诊断的情况。下图显示了色谱图中所有分析物均或多或少出现峰拖尾的现象:

第二类:仅部分分析物峰拖尾。该种情况通常表明拖尾分析物化合物和气相色谱系统的某些部分(包括色谱柱及色谱仪的相关部件等)之间存在化学相互作用,下图为该行为的典型示例:

第三类:仅溶剂峰和(或)保留时间较短的峰拖尾。此种情形亦应引起分析者注意,因为其在日常工作中时常发生,下图为溶剂峰和(或)保留时间较短的峰拖尾问题的典型情况:

第四类:仅保留时间较晚的待测物质峰拖尾。这是一类比较特殊的情况,下图为此种情况的典型图谱:
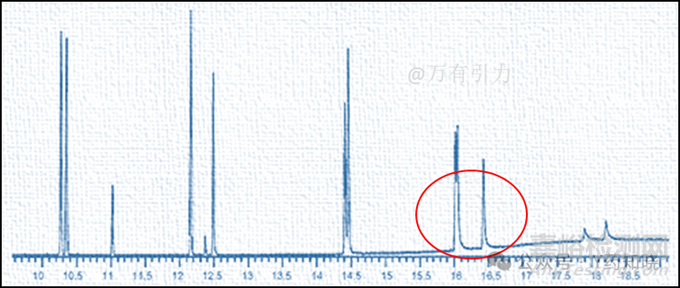
第三步解决峰拖尾
当我们通过气相色谱判断出了具体的拖尾类型以后,便可以采取相对应的解决措施去进行问题的排查和解决。
1.第一类:色谱图中所有峰均拖尾。
对第一类色谱图中所有峰均拖尾的问题,通常是由于载气在通过系统时因气路因素引起的湍流扩散引起的,也有可能由系统内未扫描的体积(或称死体积)引起的,我们可以从以下几个方面予以排查解决:
1.1检查色谱柱切面是否平整
色谱柱入口处和检测器出口处的切割质量极其重要,即使是微小的缺陷也可能导致显著的峰拖尾问题。确保色谱柱切割平滑(无锯齿状边缘),在切割过程中不产生妨碍色谱柱入口/出口的碎片,并且切割与色谱柱壁成直角。精湛的切割技术与良好的切割工具同等重要,建议由技术熟练的人员采用专业的陶瓷片及金刚石切割笔等工具进行切割。切割完成后如无不能确保切口平整,则可使用放大工具检查切口质量。使用毛刺、碎片或碎屑的锯齿状切口可引起湍流、涡流,在待测物进出色谱柱时将其截留一部分,导致峰拖尾。下图显示了色谱柱切割不佳的典型情形。

1.2查色谱柱两端安装定位是否合适
毛细管色谱柱在进样口位置是否合适通常对峰形至关重要。如果进样口内的色谱柱安装过高或过低,则分析物进入色谱柱的流路可能会卷曲或产生死体积。同样,这导致一定部分的分析物分子在进入或离开GC色谱柱时比其他分子保留更长时间,从而导致峰拖尾。虽然死体积更常与峰变宽相关,但这它可能导致峰拖尾。当然,这也适用于色谱柱在GC检测器一端的定位,因电离和非电离检测器具有较高的灵敏性,不当的安装定位也会进一步加强峰形异常。应始终严格按照仪器生产商所做的培训,将色谱柱正确的定位在进样口和检测器中,因为不同厂家、不同型号的气相色谱仪对定位的要求有所不同,但是我们只要从仪器厂商处得知明确的L1、L2(如下图所示)即可准确进行色谱柱安装定位。
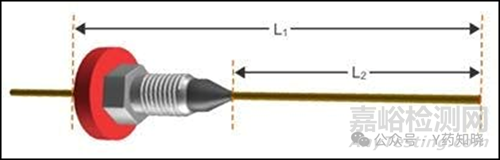
1.3检查色谱柱螺母和套圈是否使用正确
如果使用不正确的套圈进行安装,除引发除峰形问题以外可能还会伴随其他问题,如峰面积异常等。始终使用正确的套圈,包括合适的大小和材料都很重要。色谱柱安装时,柱螺母不应过紧。同样,最重要的是要严格执行仪器厂商的培训。
1.4检查色谱柱是否存在严重污染
如果色谱柱固定相涂膜受到严重污染(尤其是在入口端),则由于分析物的非理想分配和分析物非预期的严重吸附,也可能表现出拖尾行为,因为分析物组分分子无法正确分配至固定相中,相反,它们被吸附或分配到固定相表面的污染物上,这种峰拖尾的特殊情况通常伴随着峰展宽。
如果怀疑存在固定相严重污染,并且已经检查了上面提及的其他可能,则从色谱柱的入口端修剪10~20 cm,重新用样品或混标评估峰形。如果并非所有污染均已清除,则可能需要进一步切割色谱柱,以达到所需的色谱柱性能。
以上是第一类峰拖尾情况的解决思路,接下来进行第二类峰拖尾情形的解决。
2.第二类:仅部分分析物峰拖尾。
只有部分分析物峰拖尾的色谱图通常表明拖尾分析物化合物和气相色谱系统的某些部分之间存在化学相互作用。
酸性、碱性或高极性化合物易与系统内的各种组分发生二次相互作用,包括内衬、色谱柱的端面以及色谱柱内固定相被剥离的任何区域。每种极性更强的分析物的一部分分子将发生二次相互作用,通常与暴露的硅醇基团发生二次相互作用,导致大于大部分分析物分子的额外保留,因此产生峰拖尾。
硅醇基团是在硅基物质表面的极性羟基。它们通常存在于上述位置以及入口包装材料(玻璃和石英包装)的表面。这些硅醇基团取决于其构型和底层二氧化硅的性质,具有高极性,有时呈酸性,并且倾向于与极性、酸性和碱性分析物形成强氢键,导致峰拖尾。
可以采取以下若干措施来避免或纠正这些问题,包括:
2.1使用经化学灭活的内衬、GC色谱柱和配件
化学灭活可有效“封端”每个组分内的极性硅醇基团,如下图示例:
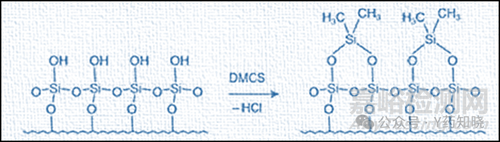
2.2选择惰性的进样口配件
仔细选择GC入口内衬管的结构材料,使其尽可能具有惰,尤其是在处理易受二次保留效应影响的极性化合物时。例如选择高度惰性的内衬管及其填充材料。
2.3定期修剪GC色谱柱.
除去暴露或剥离固定相的二氧化硅表面,并确保GC色谱柱具有最高柱效,每次使用前均检查毛刺及粗糙程度等。
2.4定期更换进样口内衬.
随着时间的推移,灭活剂将重新水解(特别是当注入含水的溶剂时),色谱柱会重新暴露内衬表面和填充材料上的硅醇基团。
需要指出的是,在预防性维护计划中采用定期的色谱柱修剪和入口衬垫维护将大大有助于通过二次保留效应减少峰拖尾的案例发生。在对单个分析物峰拖尾进行故障排除时,还有一个值得提及的现象——热分解。当热不稳定分析物在GC入口的高温环境中分解时,分解产物可能保留与分析物相似的化学性质,因此相对接近分析物峰(有时类似于峰拖尾)洗脱。当然,这种现象可能仅在样品混合物的部分色谱峰中出现。如果怀疑热分解,可以使用两种办法进行排查:
a.将进样口温度降低50℃并重新评估峰形以及整个分析的定量性能(其他分析物的挥发可能受到进样口温度降低的影响)。
b.如果初始方法使用不分流进样,则可改用小分流比(5:1或10:1),以缩短分析物在入口的停留时间,并重新评估所有目标组分的峰拖尾和分析性能,分流的引入可能会降低方法的灵敏度。
3.第三类:仅溶剂峰和(或)保留时间较短的峰拖尾
该问题几乎总是与样品进样溶剂对系统的过载有关,并且几乎仅在采用不分流进样技术时发生。在该进样模式下,进样衬管内的样品驻留时间远高于分流进样模式,并且除非进样口被“吹扫”,否则将从进样口缓慢流出,并随时间衰减。这就产生了一个严重拖尾峰的现象。可以通过优化不分流时间解决此类问题,也可以采用分流法(需关注检测灵敏度)去解决。
4.第四类:仅保留时间较晚的待测物质峰拖尾
这是一种特殊情况,通常可归因于以下两种原因中的一种:
4.1进样口温度过低
过低的进样口温度无法迅速挥发较高沸点(通常在后期洗脱)的分析物,此时适当提高进样口温度即可解决。
4.2分析物凝结
较高沸点的分析物在检测器中凝结(当使用具有加热传输线的检测器时尤其如此),此时适当提高检测器温度即可解决。
以上是对气相色谱峰拖尾问题的总结,通常我们在发现峰拖尾时,通过上述步骤一般均可解决该类问题。当然,对特殊类型化合物(如中强类的酸和碱性物质)的分离分析我们在排查的同时可以尝试具有特殊性能的色谱柱,目前市面上的一些特殊色谱柱对酸、碱性物质峰形的改良也具有较好的效果。

来源:药知晓


