您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-09-09 19:46
通过调查研究的方法,找到我国制药企业在首次工艺验证中普遍存在的问题,借助对国内外法规及相关文献研究,并结合我国制药企业的现状,提出对问题的解决策略,该研究对我国制药企业有效开展首次工艺验证有一定的指导意义。
1、前 言
2015 年 5 月 26 日,国家食品药品监督管理总局(CFDA) 发布了《药品生产质量管理规范(2010 年修订) 》的附录《确认与验证》,规定工艺验证应当包括首次验证、影响产品的重大变更后的验证、必要的再验证及在产品生命周期的持续工艺确认,以确保工艺始终处于验证状态[1]。早在 2011 年 1 月,美国食品药物管理局(FDA) 发布了《工艺验证的一般原则与惯例》指南,提出了工艺验证应贯穿药品的整个生命周期,将工艺验证分为工艺设计(Process Design) 、工艺确认(Process Qualification) 和持续工艺确认(Continued Process Verification) 三个阶段[2]。我国验证附录提到的工艺验证的“首次验证”所对应的是 FDA 所提出的工艺验证的第二个阶段: 工艺确认,该阶段包括厂房设施、设备确认和工艺性能确认两个要素,即药品从研发阶段通过技术转移至商业化生产阶段对生产工艺进行的验证,目的是确认按照研发所确定的生产工艺能够重复生产出符合预定要求的产品。笔者对国内制药企业执行首次工艺验证的情况进行了调查,发现我国制药企业在执行首次工艺验证过程中存在诸多问题,本文围绕这些问题展开相关研究,通过对国内及国际上有关工艺验证指南、法规及文献的研究,采用质量风险管理的工具及统计分析技术,提出了对我国制药企业执行首次工艺验证改进的建议。
2、我国制药企业执行首次工艺验证的现状
我国许多制药企业在进行药品首次工艺验证时,由于未经过充分有效的风险评估,验证不完善,导致在后续的商业化生产过程中不能持续、稳定的生产出合格的药品。近年来,我国制药企业在接受国内和国际认证检查中发现了诸多与工艺验证有关的缺陷,有些企业甚至被吊销 GMP 证书或者接到 FDA 的警告信或出口禁令。如 2017 年 5 月,浙江某制药企业接到美国 FDA 发出的警告信,原因是其按照所确定的工艺进行生产,有 20% 的产品出现不合格(OOS) 和超趋势(OOT) [3],说明其先前确认的工艺不能持续稳定的生产出合格的药品。
2.1 我国制药企业执行首次工艺验证的现状调查
为了解国内制药企业执行首次工艺验证的情况,笔者组织成立调查工作组,工作组成员包括注册、生产、设备、验证、检验、QA 等,对国内80 家药品生产企业执行首次工艺验证的情况进行了调查,其中包括47 家原料药供应商和 33 家制剂药下游客户,均为国内大、中型药品生产企业,其中供应商采取现场质量审计的形式,下游制剂药客户采取客户走访的形式,调查周期为 2016 年 7 月至 2017 年 7 月,调查具体内容及结果见表1。

表1 我国制药企业执行首次工艺验证情况调查表
2.2 我国药品工艺确认的现状分析
从表1 中对制药企业执行首次工艺验证情况的调查可以看出,我国制药企业在进行首次工艺验证时,很多企业还停留在传统的工艺验证的基础上,没有合理利用风险管理及统计度量工具,也没有按新的验证理念分不同阶段制定工艺验证策略,对首次工艺验证的方案设计研究不足。问题主要表现为在执行厂房设施、设备确认时未经过充分的风险评估并进行充分的确认(存在问题比例为 79%) ,在执行工艺性能确认时问题主要表现在工艺性能确认批次的确定、验证方案中生产条件的设计、取样策略及附加试验的设计和对验证的数据分析4个方面(存在问题比例分别为 94% 、75% 、70% 和60%) ,下面逐一对各项问题进行原因分析并提出相应的解决策略。
3、存在问题分析及解决策略
3.1 厂房、设施及设备确认
3.1.1 问题分析
企业存在该问题的原因一方面是由于人员理念落后,未对厂房设施、设备进行充分的风险评估,并根据风险评估的结果确定设施、设备确认的范围,没有结合具体产品工艺特性在厂房设计及设备选型时采取相应的降低风险的措施;另一方面是由于成本制约和时间限制,企业在做工程项目时强调低成本、收益快,在设施、设备选型时选择压缩资金的投入而忽视了对风险的控制,为了使研发阶段的技术成果尽快给企业带来效益,一味赶进度、压缩时间,导致没有足够的时间对设施及设备进行充分的确认。
3.1.2 解决策略
企业应按照“质量源于设计(QbD) ”的理念对厂房设施、设备进行设计,制定科学合理的项目进度表及确认计划书[4],使用风险管理工具对系统进行影响性评估,根据评估的结果,对产品质量有直接影响的系统被判定为“直接影响系统”[5],如压片机、灌装机等,被判定为对产品具有“直接影响”的系统,除了需要按照《优良的工程管理规范(GEP) 》进行设计调试之外,还需要遵循生命周期的方法按照直接影响系统验证“V”模型(图1)结合产品工艺特点、工艺参数范围及实际操作环境对系统和设备进行确认,有效的确认可以减少由于系统、设备原因所引起的生产过程变异。如在设计阶段某企业拟采购德国进口分装设备,其自带的RABS 手套工位适用于德国人操作,而在我国由于操作人员无法适应手套高度,无法进行正常的操作时的过程干预,需在设计阶段完成人员模拟操作,优化设计尺寸及工位。
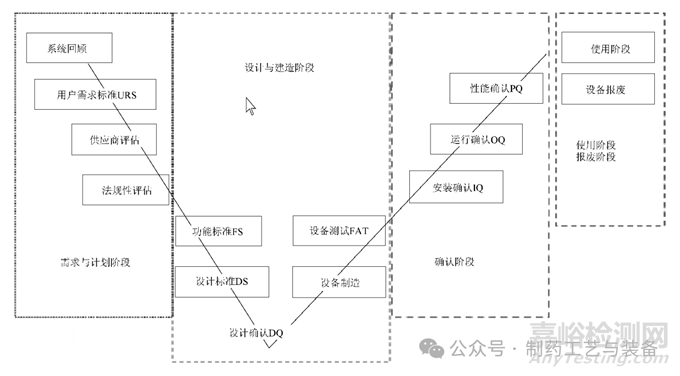
图1 直接影响系统验证“V”模型[6]
3.2 验证批次的确定
3.2.1 问题分析
根据调查结果显示,94% 的制药企业选择了三个批次的产品进行了工艺验证,由于商业化规模生产的批量、设备与研发小试阶段有较大差异,3 个批次的产品产生的数据不足以对生产工艺的可靠性进行评估[7]。产生该问题的原因一方面是因为对于验证批次是否能够上市没有法规支持,多批次验证成本高,另一方面由于大部分参与首次工艺验证的人员与产品研发阶段的人员有很大不同,参与验证人员缺乏对产品及工艺的理解,未经过充分的风险评估,仅按照传统意义上的三个批次进行了验证。
3.2.2 解决策略
应使用质量风险评估的方法确定工艺性能确认的批次,首先应对产品知识、产品工艺理解和过程控制策略三个方面可能会存在的风险因素进行评估,根据评估结果对各风险因素进行排序,将风险分为低、中、高三个等级,风险因素排序的依据是其可能导致的生产过程变异对产品安全性、有效性的影响程度及对患者造成的影响[8],表2 中分别对产品知识、产品工艺理解和过程控制策略三个方面的风险因素各举一例进行风险排序[9]。
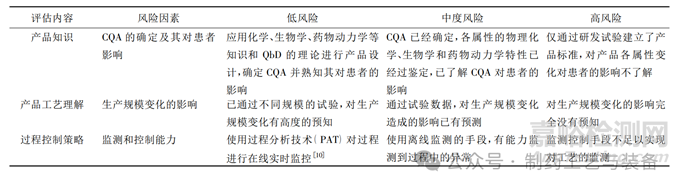
表2 制药企业风险因素排序示例
再根据对产品知识、产品工艺的理解和过程控制策略三个方面评估出的各因素高、中、低风险的情况,最终确定工艺过程总体的风险等级,根据风险等级确定需要进行的工艺确认的批次,见表3。
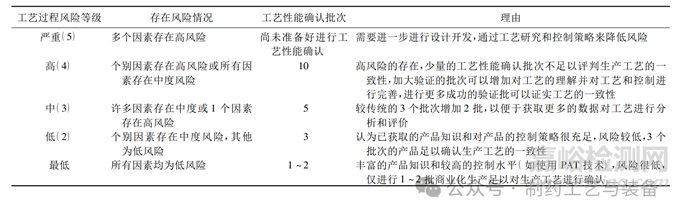
表3 根据风险等级确定工艺性能确认批次
3.3 验证方案生产条件的设计
3.3.1 问题分析
在进行工艺性能确认时,75% 的制药企业未根据实际商业化生产的产品批量、规格、设备型号等进行合理的生产条件的设计,由于产品进行技术转移时,未能将在研发阶段积累的产品及生产工艺知识有效传递至首次工艺验证阶段,验证人员对产品及工艺的理解不足,导致验证条件设计不充分,工艺性能确认不能涵盖商业化生产时所有运行条件。
3.3.2 解决策略
在进行工艺性能确认时应根据小试、中试研究结果,结合产品剂型、规格、设备型号、包装形式等,经过科学的判断和风险评估,使用合适的方法进行验证方案的设计,常见的方法有括号法 (Bracketing Approach) 、矩阵法 (Matrix Approach) 等[11],括号法适用于单个工艺变量但其他条件保持固定的情况,通过确认工艺变量的极端条件,从而确认在整个工艺控制范围内进行的操作均稳定有效,如对于某胶囊剂,有不同的装量规格,胶囊灌装前的粉碎、过筛、混合等工序的操作均一致,在进行工艺性能确认的方案设计时,可以考虑对最大灌装量和最小灌装量进行确认。矩阵法适用于在相同的工艺条件下,产品结构有多个变量的情况,如对于某一小容量注射剂生产线的生产,其产品包括3种灌装体积、4种原液浓度、5种成品规格,对其 PPQ 矩阵法设计见表4。
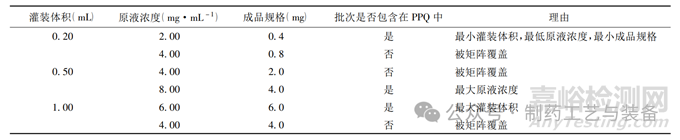
表4 矩阵法进行工艺验证生产条件设计
3.4 取样及检测策略
3.4.1 问题分析
大部分制药企业在进行工艺性能确认时,所制定的中间产品和成品的检测项目及取样频率与日常监测中规定的质量标准及取样频次相同,导致对工艺性能监测不足够,不足以获得充分的数据来评价工艺和产品质量。
3.4.2 解决策略
可根据工艺的复杂程度、对产品及工艺的理解程度或已获知的相似产品的经验进行评估,制定生产过程各个生产步骤的取样计划,包括取样点、测试项目及取样频率,取样频率的确定应结合统计学的原理,确保获得对生产工艺和产品质量进行分析和评价的足够的数据。所以,工艺性能确认的取样及检测频率应较日后持续工艺确认阶段的频率高,例如某片剂产品由多种物料混合,不易混匀,就可以在预混、总混、压片各阶段增加取样频次进行含量均匀度的检测。另外,针对于某些产品,如某抗生素原料药,在提炼过程中使用到絮凝剂,在产品检测时需要增加额外项目“絮凝剂”的检测,以确认在后续提取及精制过程中是否能够将其有效去除,而在后续的持续工艺确认过程中则不必对该项目进行检测。
3.5 对验证数据的分析
3.5.1 问题分析
在进行数据分析时,仅对中间体和成品的检验结果是否符合相应的标准进行了评价,没有利用统计分析工具进行产品批内和批间的分析,不能有效判定生产工艺是否具有重复性及过程能力是否足够,以便于采取有效的工艺改进或控制措施。
3.5.2 解决策略
可以使用统计分析工具并结合Minitab等统计分析软件实现对数据的快速分析[12],如: 对于连续型变量的批内监测(如对片重、硬度等) ,使用 X-R 或 X-S 控制图进行分析; 对于连续型变量的批次间的监测(如含量、溶出度等) ,使用 I-MR控制图进行分析; 对于关键质量属性(CQA) 和关键工艺参数(CPP) 的监测,使用 CuSum 和 EWMA 统计工具进行分析; 另外,可以使用工序能力指数(CpK)来评价工艺的控制能力水平[13]。通过使用统计分析工具对工艺过程中产生的数据进行分析,一方面确认生产工艺是否处于受控状态,另一方面可以及时发现工艺过程中的问题,及时采取改进措施。
4、对执行首次工艺验证的建议
4.1 对我国制药企业的建议
4.1.1 加强员工培训
建议制药企业引入国际先进的工艺验证理念,强化员工培训,提高员工生产过程中质量风险管控意识及对工艺确认的理解和实施水平。
4.1.2 做好产品技术转移
在产品自研发阶段向商业化生产阶段转移时,需加强转移过程的知识管理,通过做好首次工艺验证并实施过程管控,做好技术衔接[14],另外,在进行首次工艺验证时,应有研发阶段的人员参与,确保在产品研发阶段所积累的产品知识及工艺技术有效传递至商业化产品生产阶段。
4.1.3 完善设备确认
制药企业应按照设备、设施确认的要求按部就班做好各项确认工作,不要为了节约资金或赶工程进度而对后续生产造成潜在的风险。
4.2 对药监部门的建议
建议我国药政当局出台相应的有关工艺验证的指南性文件,从法规约束的角度规范我国制药企业的工艺验证,确保生产工艺持续处于验证状态。另外,建议在法规文件中对首次工艺验证批次是否“可以放行”做出明确规定,鼓励制药企业在执行首次工艺确认时,能够进行更多的生产批次以对生产工艺进行充分的确认。
5、结 语
2017 年 10 月 8 日,国家食品药品监督管理总局发布了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(以下简称“创新意见”) ,“创新意见”中对有关落实上市许可持有人法律责任的条款规定,药品上市许可持有人须确保生产工艺与批准工艺一致且生产过程持续合规,确保销售的各批次药品与申报样品质量一致[15]。在 FDA 的工艺验证指南中也提出,药品生产企业在商业化产品放行前,必须确保成功的进行了工艺确认,方可进行商业化产品的放行[2]。那如何保障 CFDA“创新意见”中所要求的“生产工艺与批准工艺一致且生产过程持续合规”呢? 那就必须进行“成功的工艺确认”。
制药企业在进行首次工艺验证时,应对厂房设施、设备按照“QbD”的理念进行良好的设计,并运用质量风险管理的方法,制定科学合理的项目进度表及确认计划书,运用验证“V”模型的方法进行确认。对工艺性能确认方案应进行基于科学的风险评估的设计,运用统计学工具对数据进行分析,以确认生产工艺可以重复生产出符合预定要求的商业化批次的产品,只有这样才可以称作进行了“成功的工艺确认”,进而再通过后续的持续工艺确认持续保障上市后药品的质量,以达到“创新意见”中所规定的“生产工艺与批准工艺一致且生产过程持续合规”的要求。首次工艺验证在产品生命周期中的验证过程中起着承上启下的作用,一方面可以发现在研发设计阶段存在的缺陷并加以改进,另一方面为后续持续工艺确认阶段的开展提供数据及技术支持,首次工艺验证的成功,是产品生命周期的一个重要的里程碑。
参考文献
[1] 国家食品药品监督管理总局 .《药品生产质量管理规范( 2010 年修订) 》附录: 确 认 与 验 证 [EB/OL]. ( 2015-05-26) [2017-10-26]. http: / /www. sda. gov. cn /WS01 /CL0087 /120500. html
[2] U. S. FDA. Guidance for industry process validation: General principles andpractices [EB/OL]. ( 2011-01) [2017-10-26] https: / /www. fda. gov /downloads/drugs/guidancecomplianceregulatoryinformation /guidances/ucm070336. pdf RegulaoryInformation /Guidances/UCM070336
[3] U. S. FDA. Warning Letters [EB/OL]. ( 2017-04-05) [2017-10-28] https: / /www. fda. gov /ICECI/EnforcementActions/WarningLetters/2017 /ucm554461. htm
[4] ICH. Pharmaceutical Development Q8( R2) [EB/OL]. ( 2009-08) [2017-11-06]. http: / /www. ich. org /fileadmin /Public_Web_Site /ICH_Products/Guidelines/Quality /Q8_R1 /Step4 /Q8_R2_Guideline. pdf
[5] ISPE. Pharmaceutical engineering guides for new and renovated facilities,Volume 5,Commissioning and qualification [EB/OL]. [2017-10-29]. http: / /www. doc88. com /p-4055459443591. html
[6] 王忠付 . 基于 V 模型的 B 企业生产设备验证管理改进研究[D]. 华南理工大学,2011.
[7] 俞育庆,张利英 . 我国药企工艺验证研究和分析[J]. 中国医药工业杂志,2017,48( 02): 284-287.
[8] ISPE. Topic1-Stage2 Process Validation: Determining and Justifying the Numberof Process Performance Qualification Batches Using Statistical Tools-Supplementto Prior Discussion Paper[EB/OL]. [2017-10-26]. http: / /www. doc88. com/p7844556673048. html
[9] ICH. Quality Risk Management Q9( R2) [EB/OL]. ( 2005-11) [2017-11-26]. http: / /www. ich. org /fileadmin /Public _ Web _ Site /ICH _ Products/Guidelines/Quality /Q9 /Step4 /Q9_Guideline. pdf
[10] EMA. Guideline on process validation for finished products-information anddata to be provided in regula submissions. [EB/OL]. ( 2016-11-21 )[2017-10-29]. http: / /www. ema. europa. eu /docs/en _ GB/document _ library /Scientific_guideline /2014 /02 /WC500162136. pdf
[11] PDA. Process Validation A Lifecycle Approach[EB/OL]. ( 2013) [2017-10-18]. http: / /www. pda. org /pcmo
[12] 王春涛,唐静,陈伟 . Minitab 软件在药品生产质量控制中的应用[J]. 中国执业药师,2012,9( 11) : 42-46.
[13] 高闪. 统计工艺控制在药品生产质量管理中的应用[EB/OL]. ( 2016-07) [2017-10-25]. http: / /www. doc88. com /p-7744592203111. html
[14] 尹美艳,赵赢 . 浅谈制剂新药从研发阶段技术转移至商业化生产时需注意的问题[J]. 医药前沿,2016. 6( 14) : 356-357.
[15] 国家食品药品监督管理总局 . 关于深化审评审批制度改革鼓励药品医疗器械 创 新 的 意 见[EB/OL]. ( 2017-10-09) [2017-11-08]. http: / /www. sda. gov. cn /WS01 /CL0050 /178289. html
本文作者贾欣秒、杨悦、赵俭,沈阳药科大学工商管理学院、华北制药股份有限公司,仅供交流学习。

来源:Internet


