您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-10-09 09:18
前言
口服给药作为一种常见且便捷的给药方式,具有较高的患者依从性。然而,口服药物的吸收是一个复杂的过程,受到多种因素的影响,包括药物的理化性质、给药剂型和胃肠道的生理环境等。
除食物可能会导致的吸收延迟、减少、增加或无影响外,口服药物的吸收还会因胃内pH值的变化而发生改变(例如多肽药物的口服生物利用度可以通过SNAC调节胃内pH值来提高),包括影响化合物在体内的溶出速率、溶解度、肠道通透性和药物的释放位置等。尤其是在大动物PK实验中,胃内pH值波动范围较大是影响实验结果均一性的重要因素。本文将介绍如何通过调控胃内pH值来优化药物吸收,并分享大动物PK实验的应对策略。
1、可乐调整胃内pH的案例
为了提升口服药物生物利用度或减少胃肠道刺激,在医嘱或药品说明书中常提到在餐前或餐后服药,然而比较特殊的情况下也有使用可乐送服的药物,即泊沙康唑。泊沙康唑是一种广谱三唑类抗真菌药物,常用于预防和治疗侵袭性真菌感染。泊沙康唑属于亲脂弱碱性化合物,其口服生物利用度会受到食物和胃酸pH的影响。研究发现,相较于空腹状态下,脱脂和高脂饮食状态下服药可以分别提高药物吸收2.6倍和4倍。然而,接受泊沙康唑治疗的患者常伴有消化系统损伤,医生通常会使用胃酸抑制剂辅助治疗,这使得患者难以通过饱腹饮食来提高药物的吸收。此外,胃内pH值的异常也会导致药物吸收的不确定性,从而影响治疗效果。因此,研究人员从胃酸pH入手,寻找改善药物吸收的办法[1]。
通过比较4种不同条件下口服泊沙康唑的效果(表1),可以看出单独使用可乐送服泊沙康唑的Cmax和AUC0-48h值更高,表明其吸收效果更好;与单独使用可乐和单独水送服相比,若联合胃酸抑制剂,则会抑制药物的吸收。
表1. 泊沙康唑血浆Cmax和AUC0-48h值(n=5) a[1]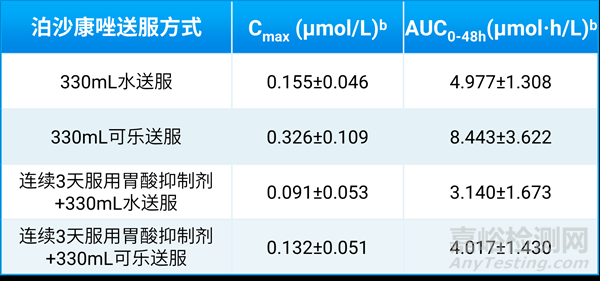
a:数值以平均值±SD表示
b:1μmol/L = 700.8 ng/mL
2、胃肠道pH对口服药物吸收的影响
由以上数据对比可知,胃肠道pH对药物的吸收会产生直接影响。
口服药物的吸收需要药物先溶解,再穿过细胞膜被人体吸收。大多数化合物为弱酸或弱碱性,具有pH依赖性溶解的特征。例如,弱碱性化合物在酸性条件下溶解度更高,而在碱性条件下溶解度较低(图1);弱酸性化合物则相反。
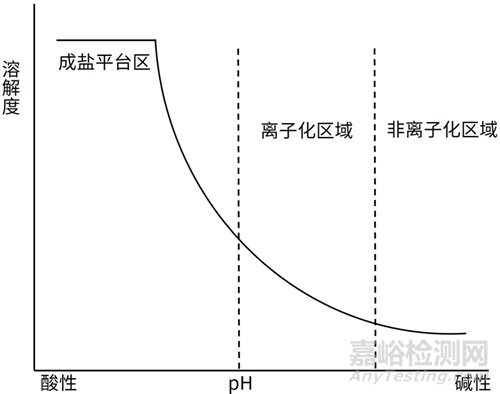
图1. 弱碱性化合物的pH溶解度曲线[2]
此外,口服药物的吸收通常以被动扩散为主,这一过程除了与药物浓度梯度、分子大小和吸收表面积有关外,还和化合物的渗透性密切相关。通常情况下,弱酸和弱碱性化合物溶解后会以离子型和非离子型形式存在。非离子型化合物亲脂性强、透膜能力强、渗透性高;而离子型化合物带有电荷,亲水性强、透膜能力弱、渗透性低。因此,非离子型化合物的比例即离子化程度决定了化合物的透膜能力。而环境pH和化合物pKa则是决定离子化程度的关键。
pKa(酸解离常数)可以理解为离子型和非离子型化合物浓度相等时的环境pH。例如,对于弱碱性化合物(pKa=9),在环境pH为9时,两者的比例为1:1。当环境pH偏碱性时,非离子型化合物占优,溶解度低,但渗透性高;当环境pH偏酸性时,离子型化合物占优,溶解度高,但渗透性低(图2)。而弱酸性化合物则与弱碱性化合物相反。
健康人群空腹时,胃酸pH值一般为1-2,而肠道pH值为5-7,口服药物在胃肠道中的环境pH经历逐渐碱化的过程。虽然胃内表面积大,但胃壁较厚且药物滞留时间较短;而肠道因小肠绒毛的存在,表面积更大且肠壁较薄,药物分子更容易透过。因此,口服药物的吸收主要表现出胃内溶解、肠道吸收的特征[4]。
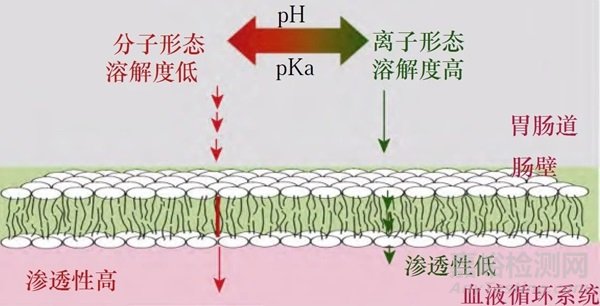
图2. 药物pH-溶解扩散模型[3]
正常情况下,弱碱性化合物在胃内的酸性环境中更易于溶解,转移至肠道后溶解度会下降;而弱酸性化合物在胃内的酸性条件下溶解度较低,转移至肠道后溶解度会增加。当胃内pH值升高时,弱碱性化合物在胃和肠道中的溶解度均会降低,从而导致体内溶出和吸收减少;而弱酸性化合物在体内的溶解和吸收则会增强。因此,在胃内pH升高的情况下,使用可乐(pH=2.5)送服泊沙康唑(弱碱性化合物),可以增加其在胃内的溶解度。此外,研究人员还发现,相较于水,可乐可以提高药物在胃内的滞留时间,进一步促进泊沙康唑的吸收。
综上所述,使用可乐送服泊沙康唑可以显著提高药物的吸收效果。因此,研究人员建议在使用该药物时,尽可能避免使用胃酸抑制剂,选择使用可乐送服[1]。
3、临床前药代动力学研究中胃pH值调整策略
口服实验动物的选择
化合物的吸收虽然可以通过体外胃肠液模拟、细胞模型和离体肠道组织模型等方法进行预测,但仍然需要结合体内动物实验的研究结果。目前,还没有数据表明有一种动物能够完全模仿人类的胃肠道特征,但通过比较不同哺乳动物的胃形态(图3)及功能,我们发现犬的胃形态与人十分接近,胃排空半衰期和其他胃肠道生理参数也与人类相似(见表2)。因此,犬作为口服给药的实验动物有更明显的优势[5]。
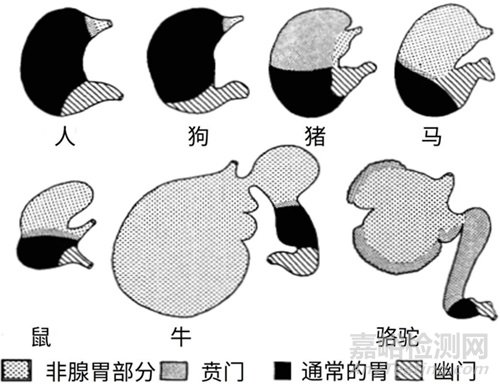
图3. 不同哺乳动物胃黏膜的类型和分布多样性[5]
表2. 不同哺乳动物胃肠道参数对比[5]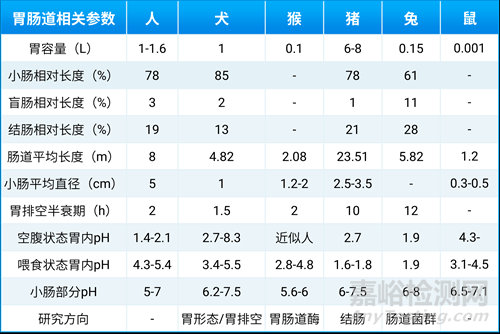
由数据对比可知,虽然猴在很多方面与人类更为接近,但实验成本较高,其口服胶囊片剂的规格也有限制。如000号胶囊规格较大,不能用于常规体型猴的口服给药;00号胶囊规格适中,但只能用于体重较大的猴,动物配合度低。而犬则没有这些限制。相比之下,猪在口服给药时动物配合度较低,连续保定和采血也更加具有挑战性;兔子的胃壁薄,无专门的功能划分且有食粪的特性,禁食情况也需额外关注,经过禁食时间测试,发现禁食24小时后,兔子胃内仍然有食糜存在。
犬胃内pH调整方法及案例
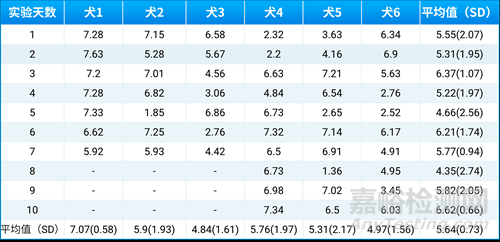
尽管犬与人类在许多方面相似,且多用于口服给药实验,但在空腹状态下,犬胃内pH波动范围较大,容易对实验结果产生影响。通过测量6只犬在10次不同实验前的胃内pH值(表3),发现空腹胃内pH平均值为5.64,最低为1.36,最高为7.63[6]。这些数据表明,同一只犬和不同犬之间的胃内pH变化范围较大,而这种变化会影响具有pH依赖性溶解的药物的吸收。在实验中经常遇到组内数据差异大、口服利用度低等问题,特别是对于依赖pH值变化进行药物释放的剂型,如肠溶胶囊,在pH值<5时稳定,而在pH值>5时释放。如果犬胃内偏碱性,可能导致肠溶胶囊提前释放,从而无法获得理想数据。
表3. 空腹犬胃内pH值变化[6]
控制犬的胃内pH水平可以通过肌肉注射五肽胃泌素和口服法莫替丁两种方法。五肽胃泌素可以刺激胃酸分泌,而法莫替丁则可以抑制胃酸分泌。
使用五肽胃泌素处理后,犬胃内pH值在20分钟内可以降至2以下,并且能够维持1小时。如果在1.5小时再次注射五肽胃泌素,则胃内pH值可以维持在2以下更长时间[6](图4)。
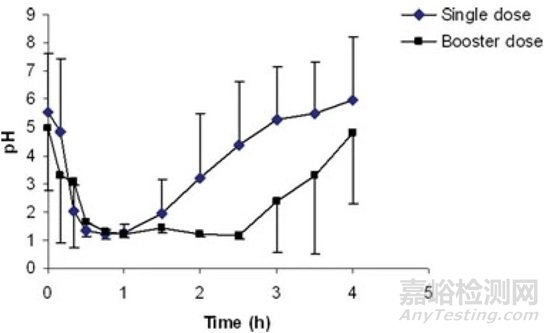
图4. 犬单次给药和初次给药后1.5小时加强剂量后的胃内pH值变化[6]
使用法莫替丁处理后,犬胃内pH值在1小时后可以达到7,并且能够维持5小时(图5)。而静脉推注法莫替丁,胃内pH值可以在10分钟后达到7以上,并维持4小时[6]。
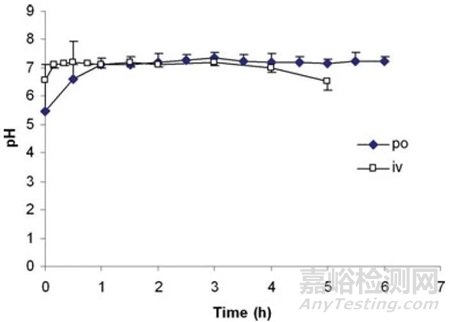
图5. 犬口服法莫替丁或静脉注射法莫替丁后胃内pH值变化[6]
为了评估pH依赖性吸收,我们进一步通过实验比较了两种不同的处理方式对药物吸收的影响。选择3只犬口服给药两种弱碱性化合物,洗脱期大于7天,分别提前肌肉注射五肽胃泌素和口服法莫替丁,其药代动力学(PK)参数比较如表4所示,对应的血浆浓度和时间曲线如图6[6]。
通过数据可以看到,两个化合物在五肽胃泌素处理条件下的Cmax和AUC值均高于法莫替丁处理,表明这两种化合物对胃内pH变化较为敏感。其中,Compound 1差异更为显著。
表4. Compound1和Compound2在五肽胃泌素和法莫替丁处理后犬体内的药动学参数[6]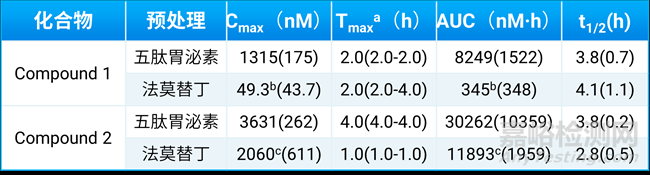
a:以中位数范围给出
b:与五肽胃泌素预处理相比,差异有统计学意义(p < 0.05)
c:与五肽胃泌素预处理相比,P = 0.07
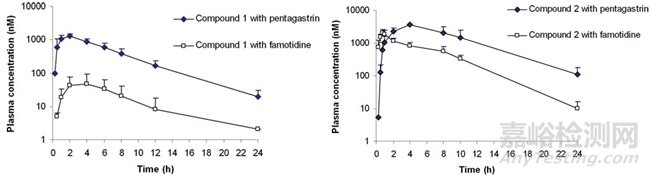
图6. 五肽胃泌素和法莫替丁预处理对Compound 1和Compound 2血浆浓度与时间曲线的影响[6]
在不同pH条件下,两个化合物的溶解度测试结果差异较大(图7)。在pH为1.1时,Compound 1溶解度最大为29mg/mL;在pH为7.0时,Compound 2的溶解度最大为38 mg/mL。随着环境pH值的升高,两个化合物的溶解度均降低。溶解度检测结果显示,随着pH值的升高,Compound 1溶解度降低比Compound 2更加显著,主要原因是Compound 1的pKa更低且对犬胃内pH的变化更为敏感。基于化合物的理化性质Pka和溶解度数据,对于溶解度低于10ug/ml的弱碱性化合物,我们可以预测其在口服吸收中受胃酸pH变化的影响,并且可以通过五肽胃泌素预处理来改善这些化合物的口服吸收[6]。
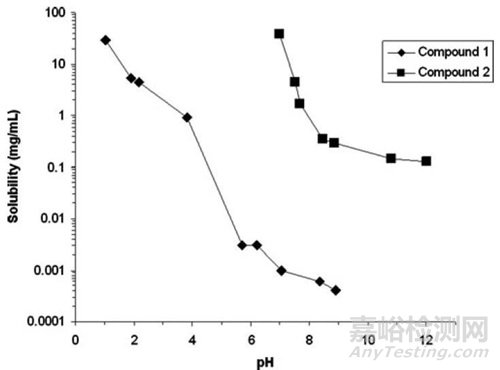
图7. pH对Compound 1和Compound 2溶解度的影响[6]
结语
通过系列实验结果可以看出,胃肠道pH的变化会影响化合物在体内的溶出速率、溶解度、肠道通透性和药物的释放位置,从而影响化合物的吸收。犬作为口服给药的实验动物有着显著的优势,但其胃内pH值波动范围较大容易影响实验结果的均一性。
为了平衡胃内pH值的波动范围,可通过对空腹犬肌注五肽胃泌素,确保犬胃内pH接近人类空腹状态水平,并保证不同犬之间胃内pH水平的一致性,从而减少因胃酸变化而造成的影响。配合口服法莫替丁,可以高效完成化合物的临床前筛选,帮助研究人员发现化合物的pH依赖性特征并积极应对,减少对胃肠道pH的影响。
参考文献:
[1] Jeroen, Walravens, Joachim, Brouwers, Isabel, Spriet et al. Effect of pH and comedication on gastrointestinal absorption of posaconazole: monitoring of intraluminal and plasma drug concentrations.[J] .Clin Pharmacokinet, 2011, 50(11): 725-734.
[2] Pallavi, Bassi,Gurpreet, Kaur,pH modulation: a mechanism to obtain pH-independent drug release.[J] .Expert Opin Drug Deliv, 2010, 7: 845-857.
[3] 郑华,郝贵周,尚萍萍,等. 基于平行人工膜渗透模型预测甲磺酸仑伐替尼胶囊生物等效性 [J]. 中国现代应用药学, 2024, 41 (13): 1775-1780.
[4] Ahmad Y, Abuhelwa,Desmond B, Williams,Richard N, Upton et al. Food, gastrointestinal pH, and models of oral drug absorption.[J] .Eur J Pharm Biopharm, 2016, 112: 234-248.
[5] T T, Kararli,Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals.[J] .Biopharm Drug Dispos, 1995, 16: 351-380.
[6] R Marcus, Fancher,Hongjian, Zhang,Bogdan, Sleczka et al. Development of a canine model to enable the preclinical assessment of pH-dependent absorption of test compounds.[J] .J Pharm Sci, 2011, 100: 2979-2988.

来源:药明康德DMPK


