您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-10-09 14:48
指南介绍
当地时间10月8日,欧盟发布MDCG 2024-11指南
指南旨在阐明哪些产品属于关于IVDR法规的范围--也称为作为体外诊断器械(IVD)或 IVD 附件的 “资格”。要获得 IVD 或 IVD 附件资格,产品必须分别符合 IVDR Art.2(2)或 Art. 2(4)中的定义。产品是否合格取决于制造商所描述的预期用途。
IVDR与关于医疗器械的MDR法规之间的界限尤为重要,因为MDR在其article 1中规定,它不适用于体外 诊断医疗器械(IVD)。
本指导文件提供了 IVD 和 IVD 附件的非详尽示例清单。更多详细示例可参阅医疗器械共同体监管框架的边界和分类手册中找到。还可参考 MDCG 2019-11。
所提供的示例是指示性的,每个制造商应根据其预期用途逐个考虑特定产品的资格。因此,检测或测量相同分析物的产品可以根据每个制造商指定的预期目的进行不同的鉴定。
1. 资格认定的一般原则
在决定产品是否属于IVDR的范围时,主要考虑IVDR Art.1和Art.2中的规定和定义。
1.1. 医疗器械和体外诊断医疗器械(IVD)的定义
IVDR Article 2 (2)将 IVD 定义为
"‘体外诊断医疗器械’是指任何试剂、试剂产品、校准品、控制材料、试剂盒、仪器、器械、设备、软件或系统,不论是单独使用还是组合使用,由制造商设计用于体外检查来自人体的标本,包括血液和组织捐献,其唯一或主要目的是提供以下一项或多项信息的医疗器械:
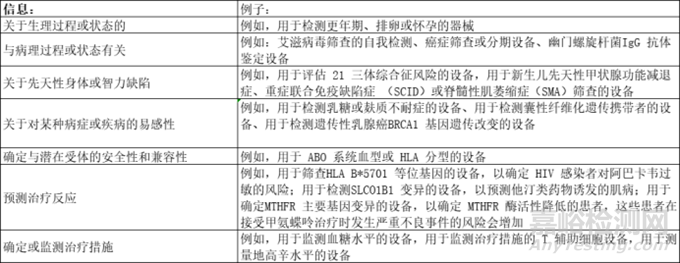
(a) 涉及生理或病理过程或状态;
(b) 关于先天性身体或精神损伤;
(c) 有关某种医疗状况或疾病的易感性;
(d) 确定潜在接受者的安全性和兼容性;
(e) 预测治疗反应或反应;
(f) 确定或监测治疗措施。
标本容器也应被视为体外诊断医疗器械"。
MDR Article 2(1)将医疗器械定义为
"‘医疗器械’是指任何仪器、器械、用具、软件、植入物、试剂、材料或其他物品,制造商打算将其单独或组合用于人类以下一个或多个特定医疗目的:
- 诊断、预防、监测、预测、预后、治疗或减轻疾病、
- 损伤或残疾的诊断、监测、治疗、缓解或补偿、
- 对解剖学或生理或病理过程或状态进行研究、替代或改造、
- 通过对来自人体(包括器官、血液和组织捐献物)的标本进行体外检查来提供信息,并且不通过药理学、免疫学或新陈代谢手段在人体内或在人体上实现其主要预期作用,但可通过这些手段辅助其功能。
下列产品也应视为医疗器械:
- 用于控制或支持受孕的器械;
- 专门用于清洁、消毒或灭菌Article 1(4)所述器械和本点第一段所述器械的产品"。
1.2 IVD 的基本特征
a) IVD 可以是试剂、试剂产品、校准品、控制材料、试剂盒、仪器、器械、设备、软件或系统。
用于诊断目的的动物,如用于检测癌症的狗,不属于 IVD。
b) IVD 可单独 使用,也可与其他装置或产品结合使用
c) IVD用于对 来自人体的标本进行体外检查 。
标本决不会再输入人体。
注:如果不涉及标本,或检查是在人体内或人体上进行(体内),则用于该检查的器械不属于 IVD(见第 2.2.2 和 2.3 节)。
d) IVD在体外 用于检查来自人体的标本。
用于检测环境或动物体内病原体的器械(例如,检测叮咬病人的蜱中的包柔氏 菌 ,以帮助诊断莱姆病)或用于兽医诊断的器械(例如,检测犬体内的 SARS-CoV-2 或癌症或奶牛体内的牛海绵状脑病)不属于 IVD。
e) IVD 的主要预期目的是为以下一种或多种医疗目的提供唯一或主要信息 :
根据IVDR Article 2(12),“预期目的 ”是指根据制造商在标签、使用说明或宣传或销售材料或声明中提供的数据,或制造商在性能评估中指定的设备用途。
一种产品可能有多种预期用途。如果这些预期目的中至少有一个是为 IVDR Article 2(2)所述的医疗目的提供信息,那么该产品必须符合 IVDR 的要求,制造商才能将其投放市场。例如,制造商打算让非专业用户了解自己的民族血统及其对某些疾病的易感性的试剂盒应被视为 IVD,因此应符合 IVDR 的规定。
无医疗用途的产品不属于 IVD。例如:用于确定祖先、寻找亲属或揭示民族血统的检测,或用于运动、健康和生活方式的检测。
注意:不能仅凭提及 IVD 或 “用于体外诊断 ”就将产品纳入 IVDR 的范围。
2. 特定资格主题
2.1. 附件
IVDR Article 1(1)规定 “本条例规定了有关 在欧盟市场上销售、在市场上提供或投入使用 供人类使用的体外诊断医疗器械及其附件的规则 ”。
IVDR Article 2(4) 将附件定义为“一种物品,虽然其本身不是 体外诊断医疗器械,但其制造商有意将其与一种或几种特定的体外诊断医疗器械一起使用 ,以专门使 体外诊断医疗器械能够按照其预期目的使用,或专门直接协助 体外诊断医疗器械实现其预期目的 的医疗功能 ”
例如:制造商专门用于特定自动 IVD 仪器的清洁溶液是该仪器的附件。
2.2. 用于采集标本的标本容器和产品
2.2.1. 标本容器
IVDR Article 2(2) 规定:
标本容器也应视为体外诊断医疗器械。
IVDR Article 2(3) 规定:
“标本容器”是指由其制造商专门用于主要(primary)密封和保存(preservation) 来自人体的标本以用于体外诊断检查的器械,无论是否为真空型。
(a)主要(primary)一词不一定是指样品在时间上的最初或第一个容器,而是指其制造商打算主要与样品直接接触并因此可能影响样品的容器
(b) “保存(preservation) ”一词并不意味着贮器必须装有标本保存剂,而是指贮器的目的是保护标本,例如防止温度波动、光照、物理破损、污染等。
在采集、运输和储存单个标本时,可能会涉及不止一个标本贮器。在这种情况下,每个贮器的制造商都必须有符合 IVDR 的证据。
例如 采血管、尿液或粪便标本容器被视为 IVD。用于储存血液以便日后输入人体的血袋被视为医疗器械,而非 IVD。
与人体直接接触的产品应按第 2.2.2.b 和 2.6.2 节所述方法处理。
其他玻璃或塑料管、杯、比色皿或在分析过程中(通过等分或其他方式)放入标本 的其他容器,不被视为 IVDR 所指的 “标本容器”,除非其制造商专门将其用于体外 诊断检查,例如作为附件。它们通常被视为一般实验室设备。
2.2.2. 用于采集标本的产品
a) 与人体无直接接触的产品
用于转移标本的产品,但其制造商并非专门为体外 诊断检查目的而对来自人 体的标本进行主要封装和保存的,不应被认定为 IVD(如将血滴从手指转移 到测量设备的塑料吸管)。
b) 与人体直接接触的产品
侵入性(MDR 所指的)标本采集装置或为获取标本而直接作用于人体的装置不得 视为体外 诊断医疗器械的附件(IVDR Article 2(4))。这些产品被视为 MDR 范围内的医疗器械。
例如:针头、柳叶刀、静脉注射器、口管、棉签、尿液收集袋、与皮肤直接接触的汗液卡,用于测量汗液中的氯离子以诊断囊性纤维化。
2.3. 不涉及标本的设备
有些诊断医疗器械不需要从病人身上提取可容纳的标本就能发挥作用。
体外诊断医疗器械的定义规定,体外诊断医疗器械 “由制造商设计用于对来自人体的标本进行体外检查”。因此,如果没有标本“来自人体” ,则该设备不应被认定为 IVD。此类产品应被认定为医疗器械。
请注意,独立软件虽然不直接分析人体标本,但如果其目的是根据其他 IVDs 的结果提供信息,也属于 IVDR 的范围(见第 2.8 节)。
举例说明:
通过能量发射(如近红外能量)在人体上检测血糖的非侵入性设备不应被认定为 IVD,而应属于医疗设备。
脉搏血氧仪通过指尖吸收红外光来测量氧/脱氧血红蛋白比值,不应作为 IVD 而应作为医疗器械。
核磁共振扫描仪不应作为 IVD 而应作为医疗器械。
2.4. 一般实验室用产品
IVDR Article 1(3)(a)规定,IVDR 不适用于:“供一般实验室使用的产品或仅供研究使用的产品,除非这些产品的制造商根据其特性特别打算将其用于体外诊断检查”。
“如果制造商根据产品的特性,明确打算将其产品用于体外 诊断检查,则该产品成为 IVD 或 IVD 附件,必须符合 IVDR 的适用要求。
但是,如果供一般实验室使用的产品不具备使其适用于一种或多种已确定的体外 诊断检查程序的具体特性,则制造商不应将该产品定为 IVD。仅在产品上添加 “体外诊断用 ”的说明不足以使产品符合 IVD 的条件。
一般实验室用产品,如石蜡、染色剂等,在体外 制备检查样本时使用,既不属于 IVD,也不属于附件,不属于 IVDR 的范围,除非此类产品的制造商根据其特性,专门将其用于体外 诊断检查或符合附件的定义。
一般实验室用产品和 IVD 产品示例(非详尽):
Article 2(2) 规定:

2.5. 仅供研究使用的产品
IVDR Recital (7)和Art.1(3)(a)规定,IVDR 不适用于仅供研究使用(RUO)的产品。
仅用于研究的产品不能由其制造商用于医疗目的(如 IVDR Article 2(2)所述)。
2.6. 同时投放市场的产品组合
2.6.1 IVD 套件
IVD 的定义包括 “试剂盒 ”本身就是一种 IVD。
IVDR Article 2(11)规定
“‘试剂盒’是指包装在一起并打算用于进行特定体外诊断检查的一套组件或其一部分”。
根据 IVDR 的规定,如果由试剂盒制造商定义的整套组件的预期用途符合 IVD 的定义,则这套包装在一起的组件可被视为试剂盒。IVD 套件本身应在外包装上打上 CE 标志,并符合 “制造商提供的信息 ”的要求(IVDR,Annex I, Chapter III.20)。
符合 IVD 定义的试剂盒可包含:
a) 仅有 IVD 和附件(如抗体、抗原、涂布 ELISA 板、标本容器),这些附件可以是:- 有 CE 标志(如果 IVD 也作为单独的设备提供);或 - 没有单独的 CE 标志;b) IVD 和以下物品的组合:- 有 CE 标志(如果 IVD 也作为单独的设备提供);或 - 没有单独的 CE 标志:
- 如果 IVDs 也作为单独的器械提供,则必须有 CE 标志;或者,
- 没有单独的 CE 标志;
b) IVDs 和以下器械的组合:
- 医疗器械(如柳叶刀、棉签),必须根据 MDR 进行 CE 标识;
- 其他产品,如一般实验室使用的产品(如用于转移病人标本的移液管);
- 食品(如为获得特定标本而添加用于诱导病人反应的口香糖)。
注:IVD 检测包可能不包括医药产品。如果一种 IVD 打算与一种药用产品一起使用,而这两种产品包装在一起,则这种组合可能不符合 IVD 套件的条件。每种产品都必须符合相应的法规。
举例说明:用于收集呼出的空气以随后检测幽门螺旋杆菌的 产品,由待摄入的标记尿素(一种药用产品)、吸管(一种医疗器械)和标本容器(一种 IVD)组成。整个共同包装的产品不能作为 IVD 试剂盒。
注:MDR Article 2(10)规定 “‘手术包’是指为用于特定医疗目的而包装在一起并投放市场的产品组合”。无论是根据MDR将其定性为 “手术包”,还是根据IVDR将其定性为 “试剂盒”,都应基于整个产品组合的预期目的。手术包的预期目的不应与 IVDR Art.2(2)相符。手术包必须至少包含一种医疗器械。如果整个产品组合被认定为手术包,则应符合 MDR 中规定的要求。手术包中包含的 IVD 部件应符合 IVDR 的要求。
2.6.2 含有医疗器械组成部分的器械
IVDR Art 1(4)规定:"任何器械,如在投放市场或投入使用时,作为一个不可分割的部分,包含了条例(欧盟)2017/745第2条第1点所定义的医疗器械,则应受该条例 [即MDR]的管辖 。本条例 [即 IVDR]的要求 应适用于 体外诊断医疗器械部分"。
MDR Art 1(7)提及了相同的概念
一个整体产品至少由两个组成部分组成,其中一个是 IVD,另一个是医疗器械,这两个组成部分通过物理方式结合在一起,形成一个物体。
例如:
- 将唾液真空吸入含有试剂材料(如用于检测艾滋病毒)的器械的集成手柄的器械,应作为医疗器械而非 IVD 合格。
- 含有综合试剂的拭子应作为医疗器械而非 IVD 合格。
- 对毛细管血液标本进行体外测量,测量间质中葡萄糖的连续体内葡萄糖监测装置应作为医疗器械而非 IVD 获得资格认证。
- 盖子上集成有棉签的标本运输管应作为医疗器械而非 IVD 合格。
下列产品不属于整体产品,即医疗器械和 IVD 组件是分开合格的,必须分别遵守 MDR 和 IVDR 的要求:
- 与 IVD 包装在一起的医疗器械,不构成一个单独的物体(见第2.6.1 节),如带有标本容器和单独拭子的标本采集包;
- 与 IVD 包装在一起的可拆卸医疗器械(见第 2.6.1 节),例如:尿液容器,可将可拆卸的漏斗固定在其上,使其紧贴身体;
- 仅在 IVD 使用说明中提及的医疗器械。
2.7. 校准品和质控品
根据Article 2(2),校准品和质控品只要符合 IVD 的定义,即可成为 IVD。
IVDR Article 2(55) and (56)规定了以下定义:“‘校准品’指用于校准器械的测量参考材料;”“‘质控品’指其制造商打算用于验证器械性能特征的物质、材料或物品;”根据Article 1(3)(c) 和(d),,国际认证的参考材料和用于外部质量评估计划的材料不视为 IVD。
如果外部质量评估(EQA)组织将这些 EQA 材料用于 IVDR Article 2(2)IVD 定义范围内的预期用途,例如作为 EQA 计划范围外的质控品或校准品,则这些材料被视为 IVD。在这种情况下,EQA 组织将被视为制造商,产品必须根据 IVDR 进行 CE 标识。
2.8. 软件
根据 IVDR Art.2(2),软件可属于 IVDR 的范围。有关软件作为 IVD 软件或医疗器械软件的进一步指导,请查阅 MDCG 2019-11。
2.9. 微生物培养基
培养基或培养基成分(如粉末状)必须由制造商专门用于提供来自人体标本的生理或病理状 态信息,才能被认定为 IVD。
根据 IVDR 的要求,培养基的此类资质包括应向用户提供的要素,特别是:
- IVD 为医疗目的提供的信息类型(微生物的存在、特征和分型);
- 说明所需的人体标本的适当类型(如血液或尿液)。
例如:含有选择剂的普通微生物培养基,如 MacConkey 琼脂和 CLED 琼脂,是否属于 IVD 取决于制造商规定的预期用途。
2.10. 染色剂
组织学、细胞学和微生物学(如涂片和组织切片)中使用的染色剂必须能提供有关人 体标本生理或病理状态的信息,才能被认定为 IVD。
根据 IVDR 的要求,染色剂的这种鉴定包括应向用户提供的要素,特别是:
- IVD 为医疗目的提供的信息类型(染色剂的特性和性能、染色剂的具体鉴定);
- 说明所需的人体标本的适当类型。
例如 Ziehl-Neelsen 染色法是否属于 IVD 取决于制造商规定的预期用途。
没有任何特定医学用途的染色剂不应被认定为 IVD。
2.11. 用于生产过程控制的检验
用于监测产品(如药用产品)生产过程的检验不应被认定为 IVD,因为它们不具有 IVDR 所述的医疗目的。
例如 用于检测血浆中传染性病原体的试验,如果是用于生产高级治疗药物产品,则不应作为IVD鉴定。
2.12. 用于生物或化学战的检验
检测环境中的生物或化学战剂的试验不应作为 IVDs,因为它们并不打算用于人体标本。
但是,如果这种试验的预期目的之一是对人体标本进行体外 检查,并具有医疗目 的,则应将其定为 IVD。
2.13. 用于执法的检验
仅用于执法过程或其它非医学目的的检验,如亲子鉴定或检测酗酒或吸毒的检验,不 应被定性为 IVD。
但是,如果以医疗为目的对人体标本进行体外 检查是某一特定产品的预期目的之一,则适用 IVDR。
2.14. 与关于生物杀灭剂产品的Regulation (EU) No 528/2012的关系
属于 IVDR 范围内的产品被排除在Regulation (EU) No 528/2012的范围之外。然而,如果杀菌剂产品属于 IVDR 的范围,并打算用于 IVDR 未涵盖的其他目的,则杀菌剂条例也适用于该杀菌剂产品,只要这些目的未被 IVDR 涉及(杀菌剂条例Article 2(2))。

来源:北京倍力医疗技术服务有


