您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-10-10 08:38
摘要:生物制品与其他药品具有明显的区别,其本身具有复杂多变的特点,对光线、温度等影响因素高度敏感,在满足普通药品稳定性有关要求的前提下,应更多的关注影响其质量的因素。生物制品在开展稳定性研究时应关注影响其质量和有效期等的因素,结合其产品特点、结构特性、质量标准制定等情况,制定适合其产品稳定性考察的方案并有效执行,控制风险。本文通过对生物制品稳定性研究的有关因素及常见缺陷分析,旨在为稳定性研究提供参考和借鉴。
稳定性研究贯穿于生物制品全生命周期,在研发阶段、支持上市阶段及上市后研究阶段中起重要的作用,是为了解生物制品原液、制剂及部分中间产品在各种环境影响下质量变化情况,是产品有效期设定的依据,可用于对产品生产工艺、制剂处方、包装材料选择及变更合理性的判断,同时也是产品质量标准制订和提升的基础。本文中生物制品是指以微生物、细胞等为起始原材料,用生物技术制成,用于预防、治疗人类疾病的无菌制剂,没有囊括所有生物制品,对于按生物制品管理等的其他特殊类别品种及新型生物制品,应根据产品的特点开展相应的研究。
本文通过对影响生物制品质量属性的关键因素进行分析与讨论,结合生物制品稳定性研究开展的各种因素、研究有关内容、不同阶段稳定性研究开展情况、近年来国内监管机构在生物制品稳定性研究方面的部分缺陷,为企业在生物制品稳定性研究开展方面提供一定参考。
1、 生物制品稳定性研究因素分析
生物制品活性成分的结构中通常含有多个活性基团,这些活性基团具有不同的化学不稳定性趋向,相比化学药品等小分子药物更易受外界环境影响,经常发生水解、酶解和氧化等化学过程,以及变性、聚集和沉淀等物理降解,对温度、湿度、光照、酸碱度等因素更敏感,可影响产品的结构特性和生物活性等质量属性,对产品质量造成不可逆影响,针对不同的稳定性研究试验需考察的试验条件有所不同[1]。
生物制品稳定性研究一般包括长期稳定性、加速稳定性和影响因素试验研究[1]。长期稳定性是生物制品研发和质量控制中的关键步骤,为制定原液和制剂成品有效期提供重要的依据,考察条件应与拟储藏条件或经批准的储藏条件保持一致,以确保药物在规定的有效期内保持其安全性、有效性和质量稳定性。加速稳定性试验通常是在比长期稳定性考察条件较高温度下进行,同时选择适宜湿度进行控制,通过加速条件考察可以评估产品在贮存、包装、运输、使用等过程中短暂偏离实际储藏条件的影响,为确定产品有效期提供数据支持,有助于阐明药物的降解途径,也可辅助于分析方法验证。影响因素试验(又称强制条件试验) 是通过强制破坏条件,了解药物分子固有的稳定性,最终确定药物的降解途径以及可能产生的降解产物,同时进一步论证分析方法的检验能力,试验条件一般比加速试验更为苛刻,在生物创新药工艺变更的质量可比性及生物类似药的分析相似性评价等方面应用较广,通过较短的考察时间,可以观察到药物质量属性的变化,便于分析不同产品之间的差异。
2、 生物制品稳定性研究有关内容
本文综述的稳定性研究包括上市申报阶段及上市后产品的稳定性研究,对于药学研究、临床阶段、相容性研究等阶段的稳定性研究可基于相应的法规或指南要求及产品特点开展。
2.1 研究样品
生物制品稳定性研究对象一般通常包括原液、成品及部分中间产品等。稳定性研究的样品批次数量应基于法规及研究目的确定,通常至少为3 批[2]。研究样品应采用与实际贮存相同的包装容器与密闭系统,或者使用相同材质的包装容器模拟包装形式,能代表规模化生产的制品,不同批次的制剂应来自不同批次的原液。对于样品浓度不一致的多种规格的产品,建议分别开展稳定性研究;对于具有不同装量、不同单位或不同重量的制剂进行稳定性试验时,一般采用矩阵法或归一法(括号法) 选择代表性样品开展稳定性研究。另外,液体制剂在稳定性研究中还应结合研究目的考虑产品放置方向,如正立、倒立或水平放置等。
2.2 研究条件
研究条件应根据研究目的和产品自身特性进行摸索与优化。根据前期研究成果对各种影响因素(如温度、湿度、光照、反复冻融、振动、氧化、酸碱等) 进行综合考虑,制定长期、加速和影响因素试验等的稳定性研究方案。
根据不同的产品特性和拟储存条件,选择合适温度进行稳定性研究。生物制品通常为冷藏或者冷冻保存,针对拟冷藏的产品建议长期考察温度为5 ℃±3 ℃,加速考察温度为(25±2)℃;针对拟冷冻存储产品, 建议长期考察温度为−(20±5)℃ , 加速考察的温度为(5±3)℃ 或(25±2)℃。对于需要在−20 ℃ 以下存储的产品,也可根据产品自身特性设定合适的试验温度。
湿度也是影响生物制品稳定性的重要因素,如果可以证明容器与密闭系统具有良好的密封性能,则不需要考虑不同湿度条件下的稳定性,同时应关注半透性容器不同容积在不同湿度条件下对产品的影响。
对于可能对光敏感的生物制品,需要进行光照条件下的稳定性,以评估光照对产品稳定性的影响。根据ICH 指导原则,样品至少需要暴露条件为总照度为1.2×106 lx·h 和近紫外灯能量为200 W·h·m−2[3]。此外, 在试验中应设置对照样品,将对照样品置于暗条件下,并置于与光照样品相同条件下,以平行比较二者之间的差异。
对于需要冷冻保存的生物制品(如原液),验证并评估其在反复冻融条件(次数基于实际生产情况) 下产品的质量变化情况;而对于冷藏保存的生物制品,尤其是原液或者中间体阶段,其是否开展需基于产品性质与实际使用情况而确定。
在实际操作中,模拟生物制品在运输和储存过程中可能遇到的振动情况,评估其对产品质量的影响,以确保其在使用过程中的安全性和有效性。
氧化是蛋白质中主要的化学降解途径之一,易于发生氧化的氨基酸残基有甲硫氨酸、色氨酸、酪氨酸和半胱氨酸,根据产品特点设计氧化试验考察其对产品质量的影响[4]。
适宜的溶液pH 值是维持蛋白质质量稳定的重要条件,在生物制品生产过程中,因工艺步骤的需要经常对中间产品进行酸碱度的调整,如高低pH 灭活等步骤,偏酸或偏碱的条件可能引起蛋白性质改变,比如多聚体的增加、片段的增多等,进而影响产品的质量。
2.3 研究项目
考虑到生物制品自身的特点,稳定性研究中应采用多种物理、化学和生物学等试验方法,对产品进行全面分析与检定。检测项目应至少包括产品敏感且可能反映产品质量、安全性和/或有效性的项目,如鉴别试验、效价、纯度等,同时建议对年度检测时间点,应尽可能进行全面检定;同时应考虑生物制品原液、制剂及中间产品在选择研究项目是应重点考察的内容。
鉴别是最直观反映产品质量和一致性的方式,结合生物制品分子结构,选取特异性高、专属性强的分析方法对产品鉴定,常规生物制品通常选取肽图、等电点或生物学等一种或两种以上方法,必要时,可采用供试品与参比品相比较;而对于新型生物制品则主要在于其核酸水平的鉴别,如CAR-T 细胞治疗产品与基因治疗产品则通常采用核酸分子鉴定技术[5]。
效价是评价生物制品稳定性的重要指标之一,直接影响到产品的疗效和安全性,根据产品作用机制,通常选择生物学活性和结合活性进行效价评估,需要与经过标准化的参比物质比较,从而获得产品的活性。
常规生物制品的纯度主要包括分子大小变异体和电荷变异体,在稳定性研究中重点关注产品的降解和聚合情况,尤其是长期稳定性试验中出现的具有明显变化的降解产物和聚合物,应尽可能进行鉴定。分子大小变异体通常采用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)、十二烷基硫酸钠-聚丙烯酰胺毛细管电泳(CE-SDS) 和分子排阻色谱(SEC) 等方法,赵健等[6] 通过对重组人源化单克隆抗体(rhumAb1) 进行高温(40℃) 处理富集分子大小变异体,结合液质联用(LCMS)的方法,对SEC 分析中的4 个组分和非还原CE-SDS 分析中的7 个组分完成结构鉴定,发现聚体为单抗的二聚体形式,主要的片段是由重链的铰链区断裂造成的。电荷变异体通常采用全柱成像毛细管等电聚焦电泳(icIEF)、离子交换色谱(IEX)、毛细管区带电泳(CZE) 等方法,Gandhi 等[7]将一种IgG1 抗体置于25 ℃ 条件下,孵育6个月,针对稳定性过程中显著增加的酸性组分开展了详细的研究,结果表明酸性组分的增加主要是由蛋白质的脱酰胺、异构化和片段等降解途径引起的。
不溶性微粒也是评价产品,尤其是成品(注射剂) 安全性的重要指标之一,对于注射液中不溶性微粒检查方法,各国药典收载的方法均为光阻法和显微计数法,但是对各方法的适用条件的限定不同,企业应基于产品特性,制定科学合理的控制措施[8]。对于生物制品注射剂而言,产品中的蛋白有聚集并形成不溶性微粒的倾向,且这种聚集会在一定程度上影响药效,增加潜在的免疫原性风险,因此不溶性微粒检测也应包括在稳定性考察中。
另外,外观(例如颜色和澄清度等)、含量、可见异物、pH 值、注射用无菌粉末的水分和无菌检查等项目应结合产品特性开展相应研究。对于一些特殊的生物制品,其稳定性考察研究的项目应基于其分子结构或者特性开展相应的研究[9],如抗体偶联药物(antibody drug conjugates, ADCs)作为一类特殊的抗体药物,是由抗体、小分子和连接子三部分组成, 不同的偶联方式产生的ADC 分子形式不同, 药物/抗体比率(drug to antibody ratio, DAR) 也会有所不同[10], DAR 值是ADC 药物稳定性的关键评价指标,应结合分子特性开发适宜的检测方法, 常用的方法包括HPLC/UV、LC-QTOF/MS 和UV-Vis 等[11],尽管小分子通常采用共价结合的方式与抗体进行偶联,但是小分子的脱落也是在稳定性研究中值得关注的[12]。
2.4 研究时间
生物制品稳定性研究时间根据产品的有效期及放置条件的不同,设定的研究时间点有所差异,在实际操作过程中根据产品质量情况,比如初期稳定性试验结果,灵活调整时间安排。
长期稳定性时间点根据ICH[13] 和中国药典中稳定性试验指导原则有关要求,如果产品的预定有效期在1 年或更短,那么在前3 个月,建议每月进行1 次稳定性试验,之后则每3 个月进行1 次。若产品的有效期超过1 年,则在第1 年内每3 个月进行1 次试验,第2 年每6 个月进行1 次,之后每年进行1 次。长期稳定性试验的目的是确保产品在整个有效期内始终符合标准要求,原则上应该在《药品生产质量管理规范》(Good Manufacturing Practice of Medical Products,GMP) 规定的存放时限内持续进行,直至产品被判定为不合格为止。
加速稳定性一般是介于长期稳定性试验和影响因素试验之间,在加速稳定性为期考察6 个月的研究中,至少在开始和结束等的3 个时间点进行考察,必要时可增加考察频率,应尽可能观察到产品不合格。
影响因素是比加速试验更加剧烈的条件,有助于识别产品可能的降解产物,研究的持续时间通常达到产品发生5%~20% 的降解,保证降解产物的水平是药物的真实降解而不是方法变异,同时避免药物发生过度降解[14]。
3、 不同阶段稳定性研究开展情况
稳定性试验是贯穿于整个药品研制(研发、临床)、上市及上市后质量研究的各个阶段,如何根据不同阶段的目的和特点,科学、有效、高效地设计和开展稳定性研究非常的重要。
3.1 药品研制阶段
在早期研发阶段,往往对产品特征尚缺乏全面的了解,工艺和控制尚未确定和优化,为支持产品通过新药临床研究申请(investigational new drug,IND),通常选择合理且科学的稳定性研究策略。IND 申报时需提供工艺、生产、质量、动物药理学/毒理学以及临床方案等信息,以评估该产品安全性是否支持进入人体试验。在《新药Ⅰ期临床试验申请技术指南》中指出“稳定性研究结果应能支持I 期临床试验”[15] ,申报临床时,临床试验用的药物原液和制剂应进行长期和加速稳定性研究,制剂长期稳定性考察时长应涵盖药物放行检测、储存和运输、早期临床试验方案中给药时间和间隔周期等[16]。由于生物制品通常具有较复杂的结构,因此在临床前研究阶段,也应适当进行影响因素稳定性研究[17],有助于对产品特性进行评估,方便后续临床试验用药物生产、保存和运输条件的确定。此外,还应考虑临床用药的给药方式,进行相应的配伍稳定性试验,已确保临床用药的安全性及有效性。
3.2 支持药品上市阶段
根据《临床试验期间生物制品药学研究和变更技术指导原则》(征求意见稿)(2023 年7 月),确证性临床试验阶段,应参考相关指导原则,制定全面的稳定性研究的方案,以支持拟申请上市产品贮存期的设定[18]。
产品有效期的制定应根据真实时间/真实温度的数据,因此应选择≥3 批能代表生产规模情况的原液和制剂进行长期条件下的研究,稳定性研究的期限应至少能够涵盖所开展的临床试验的要求[3]。此外,应在上市申请前完成原液和制剂的加速条件和影响因素试验(如极端pH、光照、振荡、冻融、高温、氧化等),以确认原液和制剂的内在的稳定性、潜在的降解途径及拟用的分析方法的稳定性指示能力与适用性,为产品有效期的制定提供支持性证据。对于产品的容器包装系统,在临床试验期间应开展制剂贮存容器全面的相容性(浸出物、可提取物)、密封性研究,已确认所使用包装系统的安全性[18]。此外,生物制品通常要求冷链保存和运输,应对产品的运输条件开展模拟试验研究。在稳定性试验时,充分考虑运输路线、交通工具、运输距离、运输时间、装载模式、外界环境以及运输时可能遇到的最差条件,确认产品在运输过程中处于拟定的保存条件下可以保持产品的稳定性,并评估产品在短暂地脱离拟定保存条件下对产品质量的影响,尽可能对产品脱离冷链的温度、次数、总时间等制定相应的要求[13]。
若在临床试验期间产生药学变更,应按需提供强制/加速稳定性对比研究数据以及长期稳定性数据,提供变更后稳定性研究计划,并承诺按照稳定性研究计划进行稳定性研究。若涉及,还应提供直接接触包装材料的相容性研究数据[18]。
3.3 上市后研究阶段
药品上市后,仍然需要对商业化生产阶段的药品进行稳定性研究,通常前期研究阶段的稳定性都具有一定的局限性,上市产品的真实稳定性,应采用实际生产条件下商业化规模的样品进行考察。上市后稳定性研究主要包括持续稳定性研究和变更/偏差等调查的稳定性研究。持续稳定性研究包括加速试验和长期试验,目的是在有效期内监控已上市药品的质量,对药品的包装、贮存条件和有效期进行进一步的确认。
若生物制品在获得上市许可后,在生产、质控方面发生变更,则需对变更前后产品开展稳定性可比性分析,评价变更对产品质量的影响。变更引发的稳定性研究方案需要根据变更对产品的影响程度综合确定,需能充分反映变更后药品的稳定性变化情况,可通过进行加速或者强制降解稳定性试验来确定产品的降解趋势,对比变更前后的产品质量。如果原液的变更可能会影响制剂的稳定性,则应同时对原液和制剂进行强制降解和/或加速稳定性可比研究和长期稳定性考察[19]。
4、 生物制品核查稳定性研究方面中常见缺陷举例
通过对近年来在生物制品注册核查及日常检查过程中有关缺陷进行汇总与分析,发现主要集中在稳定性试验方案制定与执行方面、数据可靠性(异常结果处置)、检验过程及相关记录规范性等方面,与化学药品注册核查稳定性研究常见缺陷类似[20],见表1。
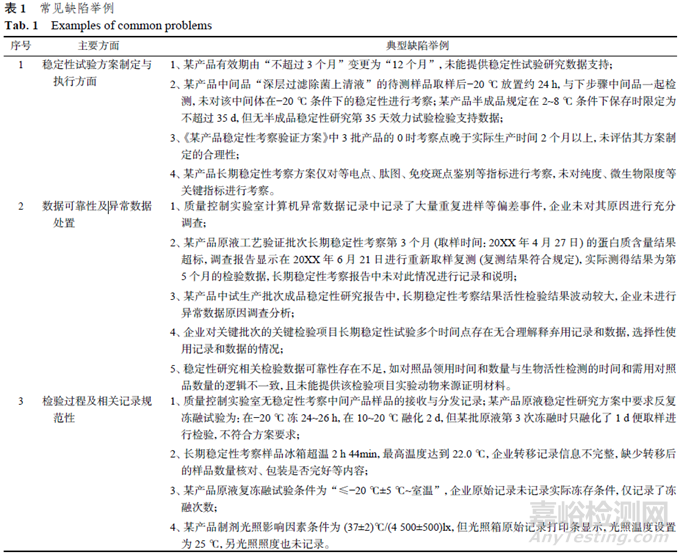
5、 分析与探讨
药品稳定性研究贯穿于制品的全生命周期中,对于评价产品质量和疗效具有非常重要的意义。通过系统的稳定性研究以全面考察其质量和疗效的变化,探究分子固有稳定性,为药品有效期的制定及后续生产、储存、使用等条件提供充分的依据。
在稳定性方案制定和执行方面,充分评估产品特性及各个试验因素的影响,制定科学合理的方案,按照方案设置相应条件并彻底的执行;在数据可靠性及异常数据的处置方面,建立合理的接受标准和检验方法,必要时采用适宜的统计方法对试验结果进行评估,有些数据在稳定性研究过程变异较小,通常无需对此进行统计学分析,只需提供简单的理由即可。对于不同批次产品的稳定性数据应评估批间的一致性,同一批次的产品在不同时间点的稳定性数据应进行趋势分析,对于发现的异常数据及时、合理的进行相应的调查处置;在检验过程规范性方面按照GMP 中质量控制实验的有关要求开展,规范影响的检验操作及记录。
同时,在产品的不同阶段,需根据各阶段的特点和目的,采用不同的稳定性考察手段,以期能更好的完成各阶段的研究目标,对不同阶段的产品稳定性要求不能一概而论,如在药品研制阶段,尤其是早期研发阶段,其制剂稳定性的目的主要用于探索不同处方下药物的稳定性,试验设计中会保留变动的空间,已达到试验目的;另外,在研发阶段也应考虑制品、辅料等与包材相互作用,如包材中硅油对于制剂稳定性的影响,是否有辅料的降解作用等,都应予以关注;而以上市申请阶段的目的则更多的是验证性试验,试验条件准备足够充分,方案设计足够完善,以证明产品的稳定性可满足申报要求;产品上市后进行的稳定性试验属于是监控性试验,用于监控已上市产品在效期内的稳定性,保证已上市产品的质量。
生物制品生产企业或者持有人在开展稳定性研究时,结合生物制品自身产品特点及质量管理体系建设情况,在全生命周期的不同阶段,对影响产品质量和疗效的各关键因素进行充分评估,切实落实稳定性方案的执行,关注研究过程中出现的异常结果或趋势,不断地提升稳定性研究水平,切实保障药品的安全。
参考文献
[1] MU M, LIU B Y, LIANG W Y. Brief introduction of guideline for stability test of biological products in the Chinese Pharmacopoeia 2020[J]. Drug Stand China(中国药品标准),2022, 23(2): 144-147.
[2] ICH Q5C, Quality of biotechnological products: Stability testing of biotechnological/biological products[EB/OL]. (1995-12-30)[2023-12-26]. https://database.ich.org/sites/default/files/Q5C%20Guideline.pdf.
[3] ICH Q1B, Stability testing: Photostability testing of new drug substances and products[EB/OL].(1996-11-6)[2023-12-26].https://database.ich.org/sites/default/files/Q1B%20Guideline.pdf.
[4] 国家药品监督管理局药品审评中心. 生物制品稳定性研究技术 指 导 原 则 (试 行 )[EB/OL].(2015-4-15)[2024-1-3].https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=f1589779e6723f65431662d0a4dea9d6.
[5] SHI X C, QIN X, YUE L, et al. General technical requirementsfor the quality control of gene therapy products[J]. Drug Stand China(中国药品标准), 2022, 23(3): 229-235.
[6] ZHAO J, WU Z H, LÜ M, et al. Characterization of the size variants of a recombinant humanized monoclonal antibody(rhumAb1)[J]. Acta Pharm Sin(药 学 学 报 ), 2016,51(12): 1897-1905.
[7] GANDHI S, REN D, XIAO G, et al. Elucidation of degradants in acidic peak of cation exchange chromatography in an IgG1 monoclonal antibody formed on long-term storage in a liquid formulation[J]. Pharm Res, 2012, 29(1): 209-224.
[8] LI Y, SHA X Y. Regulatory requirements of sub-visible particles for injections in different countries and its application in biotech drugs[J]. Chin Pharm Aff(中国药事), 2024, 38(1):11-23.
[9] TONG H J, LIU B. Research progress on the chemical methods for site-specific coupling of antibodies and drugs[J]. Chin J Mod Appl Pharm(中国现代应用药学),2022, 39(22): 3030-3037.
[10] TSUCHIKAMA K, AN Z Q. Antibody-drug conjugates:Recent advances in conjugation and linker chemistries[J].Protein Cell, 2018, 9(1): 33-46.
[11] MATSUDA Y, MENDELSOHN B A. Recent advances in drug-antibody ratio determination of antibody-drug conjugates[J]. Chem Pharm Bull, 2021, 69(10): 976-983.
[12] ROSS P L, WOLFE J L. Physical and chemical stability of antibody drug conjugates: Current status[J]. J Pharm Sci, 2016,105(2): 391-397.
[13] ICH Q1A, Stability testing of new drug substances and products[EB/OL]. (2003-2-6)[2024-1-15].https://database.ich.org/sites/default/files/Q1A%28R2%29%20Guideline.pdf.
[14] CHAN C P. Forced degradation studies: Current trends and future perspectives for protein-based therapeutics[J]. Expert Rev Proteomics, 2016, 13(7): 651-658.
[15] 国家药品监督管理局. 新药Ⅰ期临床试验申请技术指南( 2018 年第16 号)[EB/OL]. (2018-1-25)[2024-1-25].https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20180125135801903.html.
[16] SONG L N, CUI J, HE W. General Chemistry Manufacture and Control (CMC) issues during Investigational New Drug(IND) application of innovative monoclonal antibody products[J]. Chin J Clin Pharmacol(中国临床药理学杂志),2021, 37(10): 1301-1304.
[17] 刘倩, 雷利芳. 生物制品稳定性研究综述[C]//中国药学会第二届药物检测质量管理学术研讨会论文集. 西安, 2015: 170-173.
[18] 国家药品监督管理局药品审评中心. 临床试验期间生物制品药学研究和变更技术指导原则(征求意见稿)[EB/OL].(2024-6-7)[2024-9-13].https://www.cde.org.cn/main/news/viewInfoCommon/4df52a1-1c448aef5294d18f53c53d068.
[19] 国家药品监督管理局药品审评中心. 已上市生物制品药学变更 研 究 技 术 指 导 原 则 (试 行 )(2021 年 第 31 号 )[EB/OL].(2021-6-25)[2024-2-4].https://www.cde.org.cn/main/news/viewInfoCommon/7ef3a0d630aea8a49186f49f31a6fd3c.
[20] ZHOU M M, JIANG F, HUANG Y. Relevant requirements and common defect analysis for stability research of drug[J]. Chin Pharm Aff(中国药事), 2023, 37(11): 1282-1289.

来源:Internet


