您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-10-12 08:42
在庆祝2024年国庆佳节前夕,国家药典委员会于9月30日贴心献上了一份特别的“大礼包”。其在官方网站上正式发布了“关于《药包材无菌和微生物限度检查法》标准草案的公示”,标志着该标准草案正式进入公众视野,并拟将其纳入即将出版的2025年版《中国药典》中,方法编号为4401。此次公示自发布之日起,为期一个月。
该指导原则以风险管理的核心理念为基石,为药包材成品在质量控制环节中涉及的微生物检测项目提供了全面而细致的指导。它不仅涵盖了检测项目的设置、检测方法的建立和验证等关键要素,旨在满足药包材生产企业对于质量标准制定和微生物控制的迫切需求。
今天,小编将带领大家深入学习并探讨这一重要标准——药包材的微生物限度检查和无菌检查,以期帮助大家更好地理解和应用这一新标准,共同推动药包材行业的健康发展。
1、为什么药包材需要进行微生物控制?
药包材微生物控制对于确保药品的安全性、有效性和质量具有举足轻重的地位。尽管药包材如塑料、玻璃等材料本身不易成为微生物滋生的温床,且热加工等工艺能有效降低初始生物负载,但严格的微生物控制仍是不可或缺的一环。在生产、储存和使用过程中,微生物污染可能随时侵入药包材,进而影响药品质量。
为确保药品质量安全,药包材微生物限度检查和无菌检查成为至关重要的环节。这些检查能够保障药包材在有效期内质量稳定,有效防止微生物污染。制定微生物检查标准的核心目的在于预防污染,确保药品的安全性和有效性,这些标准应与药品工艺紧密结合,充分考虑微生物污染和增殖风险及其对药品质量的影响。
在标准制定过程中,质量风险管理理念至关重要,它确保微生物控制的科学性和合理性。这包括对药包材生产过程中的原料、中间品、成品以及生产环境、设备进行全面的微生物检测和监测。对于特殊药包材,如共挤膜加工成的输液袋,药品生产企业还需对加工工艺可能带来的微生物负载水平变化进行合理评估。
综上所述,药包材微生物检查标准的制定是一个涉及全生命周期微生物控制的全面过程,旨在科学合理地控制微生物污染风险,提高药品的质量保证水平,确保药品的安全性和有效性。
2、药包材微生物检测该检测什么?
药包材生产企业应基于风险评估对微生物污染进行合理控制,避免失控及过度控制情况的出现,根据药包材的不同用途制定检测项目,下面我们了解微生物限度检查和无菌检查的定义。
2.1 什么是无菌检查?
无菌检查法系用于检查药典要求无菌的药品、生物制品、医疗器具、原料、辅料及其他品种是否无菌的一种方法。药包材无菌检查法系用于检查药包材是否无菌的一种方法。若供试品符合无菌检查法的规定,仅表明供试品在该检验条件下未发现微生物污染。
药包材无菌检查法是确保药包材不含有存活微生物的重要检测手段。这种检查方法遵循无菌检查法(通则1101),涵盖了试验环境、培养基选择、稀释液、冲洗液、方法适用性试验、培养条件以及结果的观察和判断。检验数量通常与医疗器械的无菌检查标准一致。
由于无菌药包材直接参与药品的无菌生产,因此需要对内外表面进行彻底的无菌检查。药包材的无菌检查可能包括将供试品拆散或切碎后接种到培养基中,或者对于塑料瓶、软袋等,通过充分冲洗后合并冲洗液进行无菌检查。这些步骤确保了药包材在药品生产过程中不会成为微生物污染的来源。
2.2 什么是微生物限度检查?
微生物限度检查法系检查非规定灭菌制剂及其原料、辅料受微生物污染程度的方法。检查项目包括需氧菌总数、霉菌和酵母菌总数及控制菌检查。药包材微生物限度检查法包括微生物计数法及控制菌检查法。微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数,包括需氧菌总数、霉菌和酵母菌总数测定。控制菌检查法系用于在规定的试验条件下,检查供试品中是否存在特定的微生物。
药包材微生物限度检查是确保药包材在非无菌状态下微生物污染控制在安全范围内的重要步骤。这一检查应遵循非无菌产品微生物限度检查的两个通则:微生物计数法(通则1105)和控制菌检查法(通则1106),药包材微生物限度检查要点摘录如下表:
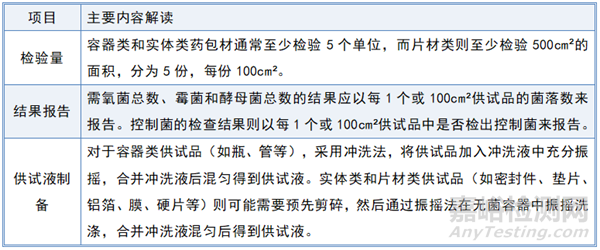
2.3 药包材成品检测项目和检测频次
药包材的微生物控制是确保药品安全的关键环节,但由于药包材种类繁多,材质和形态各异,因此无法为每个品种定制具体的检测方法和参数。
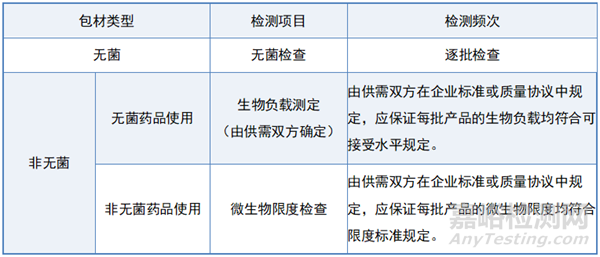
无菌药包材:由于直接参与药品的无菌生产,因此需要进行逐批的无菌检查,确保产品在生产过程中未受到微生物污染。
非无菌药包材:需要根据药包材的用途和药品生产工艺的特点,定期进行生物负载测定和微生物限度检查,以评估和控制微生物污染的风险。
检测频次应基于对药包材生产过程的风险评估来确定,以确保每批产品在微生物控制方面符合预定的质量标准。通过这样的质量控制措施,可以确保药品的安全性和有效性。
药包材成品的检测项目、检测频次如下表所示:
表-药包材成品检测项目、检测频次
参考文献
[1]www.chp.org.cn

来源:注册圈


