您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-10-14 09:05
随着监管方对药品理解的深入,对临床试验的要求也更科学,2024年9月国家药品监督管理局药品审评中心发布了ICH《M13A: 口服固体速释制剂的生物等效性》,其中对于非高风险的口服速释制剂,法规明确可进行单个空腹条件下的研究以证明生物等效性。本文以此为出发点,对基于BSC分类的生物等效性豁免或简化的相关指导原则进行分析。
一、生物等效性豁免相关法规
在仿制药的开发中,自制制剂和参比制剂同时达到药学等效和临床等效才能确保患者用药的安全、可控和有效性。生物等效性(BE)是指在相同的试验条件下,单次或多次给予相同剂量的试验药物后,受试制剂中药物的吸收速度和吸收程度与参比制剂的差异在可接受范围内。研究生物等效性的方法有多种,根据每种方法的评价效力,主要分为:(1)药代动力学研究(PK);(2)药效动力学研究(PD),在药动学研究方法不适用的情况下,可采用经过验证的药效动力学研究方法进行生物等效性研究 ;(3)临床研究,当上述方法均不适用时,可采用以患者临床疗效为终点评价指标的临床研究方法验证生物等效性;(4)体外研究(如:碳酸镧咀嚼片、碳酸思维拉姆片),体外研究仅适用于特殊情况,例如在肠道内结合胆汁酸的药物等。对于进入循环系统起效的药物,不推荐采用体外研究的方法评价等效性。
国内外关于生物等效性豁免的相关重点法规汇总如下:
|
WHO: 2006年发布《仿制药:建立可替代性的注册要求指导原则》及豁免名单。 Guidelines on registration requirements to establish interchangeability .(2006) 2015 年发布新版《仿制药:建立可替代性的注册要求指导原则》。 Guidelines RegistrationRequirements EstablishInterchangeability. (2015) |
|
EMA: 2010年发布了关于生物利用度的指导说明 Note for guidance on the investigation of bioavailability and bioequivalence CPMP/EWP/QWP/1401/98Rev1, AppendixIII [S]. 2010 |
|
FDA: 2000年FDA 发布了免除体内生物等效性试验的行业指导原则。 Guidance for industry: Waiver of in vivo bioavailability and bioequivalence studies for immediate - release solid oral dosage forms based on a Biopharmaceutics Classification System [S]. 2017发布了根据BCS豁免速释固体口服制剂体内生物利用度和生物等效性研究指导原则正式版本 Guidance for industry: waiver of in vivo bioavailability and aastudies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system [EB/OL]. |
|
ICH: M9:《基于生物药剂学分类系统的生物等效性豁免》(2019.11.20终版发布)。 M9相关问答(2019.11.20终版发布)。 |
|
NMPA: 2016年国家局发布了《人体生物等效性试验豁免指导原则》 2018年国家药品监督管理局发布了《可豁免或简化人体生物等效性(BE)试验品种》的通告。 2018年国家药品监督管理局发布了《可豁免或简化人体生物等效性(BE)试验品种(第二批) 》的通告。 2019年一致性评价办公室发了布《国内特有品种评价建议》的通知。 2019年关于发布部分新注册分类化学仿制药可参照《人体生物等效性豁免指导原则》进行研发与技术审评的通知 |
二、基于BCS分类的生物等效性豁免
上述各国生物等效性的相关法规中描述的豁免主要应用于药品从临床开发阶段转至商业化生产阶段的产品比较、仿制药的上市申请和药品上市后的变更申请。豁免的类型主要有三种:(1)基于BCS分类的生物等效性豁免;(2)基于多规格的生物等效性豁免;(3)其他可豁免生物等效性的药物。本文重点阐述法规对基于BCS分类的生物等效性豁免的相关事项和其他一些特殊的豁免类型。
2.1溶解性渗透性研究
BCS分类法是根据药物的溶解性和渗透性对药物进行分类,BCSⅠ类为高溶解高渗透性药物、BCSⅡ类为低溶解高渗透性药物、BCSⅢ类为高溶解低渗透性药物、BCSⅣ则为低溶解低渗透性药物,详见以下示意图。基于BCS分类的生物等效性豁免仅适用于BCSⅠ和BCSⅢ类药物的普通口服固体剂型和混悬剂,但不适用于窄治疗窗的药物制剂(如:地高辛、锂制剂、苯妥英、茶碱和华、法林阻凝剂等)。
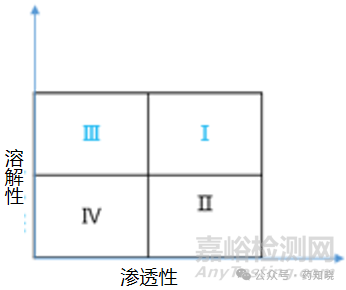
需要注意的是,目前国内对基于BCS分类的生物等效性豁免的要求,明确了不包含口腔内吸收的剂型,如含片和舌下片。但未提及是否必须为发挥全身治疗作用的制剂,也未提及口腔崩解片、分散片和复方制剂是否适用。当遇到此类情况,需要咨询监管机构并做进一步科学评估。
表1 各国法规的适用范围
|
项目 |
FDA |
WHO |
EMA |
ICH |
NMPA |
|
剂型 |
口服固体速释制剂 |
同FDA |
同FDA |
同FDA |
同FDA |
|
提及 |
不包括口腔内吸收的剂型,如含片和舌下片 |
复方制剂按“最差条件”进行试验 |
发挥全身治疗作用的制剂; 对于口腔分散片或口腔崩解片,应排除口腔吸收; 适用于速释型复方制剂,条件是复方中的所有活性成分都属于BCS 分类Ⅰ或Ⅲ,并且辅料符合章节IV.2 的要求 |
发挥全身作用的制剂;仅适用于用水送服的给药方式;适用于速释型复方制剂,条件是复方中的所有活性成分都属于BCS 分类Ⅰ或Ⅲ,且均符合第2章和第3章的标准。 |
同FDA |
|
未提及 |
是否必须为发挥全身治疗作用的制剂; 口腔崩解片、分散片的适用性; 复方制剂的适用性 |
是否必须为发挥全身治疗作用的制剂; 口腔内吸收的剂型的适用性,如含片和舌下片; 口腔崩解片、分散片的适用性。 |
— |
— |
同FDA |
在对BCS的分类及各国法规对基于BCS分类的生物等效性豁免有初步的了解后,需要我们药品研发人员真正理解各国法规中对高溶解性和高渗透性的具体实施要求,切勿与各国药典中对药物溶解性的定义混淆,具体见表2。
表2 各国法规对高溶解性药物的要求
|
项目 |
FDA |
WHO |
EMA |
ICH |
NMPA |
|
高溶解性 |
单次给药的速释制剂最高剂量能在≤250mLpH值在1.0~6.8、37±1℃的水溶性介质中完全溶解 |
单次治疗的最高剂量能在≤250mLpH值在1.2~6.8、37±1℃的水溶性介质中完全溶解 |
单次给药的速释制剂最高剂量能在≤250mLpH值在1.0~6.8、37±1℃的水溶性介质中完全溶解 |
单次治疗的最高剂量能在≤250mLpH值在1.2~6.8、37±1℃的水溶性介质中完全溶解 |
同FDA |
|
测定 |
API在37±1℃、pH1.0~6.8 水溶液中pH-溶解度曲线 |
API在37±1℃、pH1.2~6.8 水溶液中pH-溶解度曲线 |
API在37±1℃、pH1.0~6.8 水溶液中pH-溶解度曲线 |
API在37±1℃、pH1.2~6.8 水溶液中pH-溶解度曲线 |
同FDA |
|
方法 |
摇瓶法和酸碱滴定法或其他 |
— |
摇瓶法或其他 |
摇瓶技术或其他可替代的方法 |
同FDA |
|
pH值 |
根据API的解离常数来确定,包括pH=pKa、pH=pKa+1、pH=pKa-1、pH=1.0和6.8 这几个点 |
—
|
应至少在该范围内的3份缓冲液中进行研究(pH值最好为1.2、4.5和6.8),并且如果在规定的pH值范围内,还应在pKa条件下进行研究 |
同EMA,应在添加API后和平衡溶解度研究结束时测定每种试验溶液的pH值,以确保溶解度测定是在指定pH值下进行 |
同FDA
|
|
次数 |
平行测定3次或重复测定 |
至少平行测定3次 |
重复测定 |
至少3次重复定 |
同FDA |
目前国内对高溶解性药物的要求是最高剂量的API在小于等于250ml( pH1.0~6.8 )的介质中溶解,在实际测定过程中需要注意pH值至少包括pKa、pKa+1、pKa-1、1.0和6.8 这几个值,并应在添加API后和结束时分别测定pH值,必要时调节pH,平行测定3份。结果要以pH1.0~6.8范围内测得的最低溶解度对API进行分类。
表3 各国法规对高渗透性药物的要求
|
项目 |
FDA |
WHO |
EMA |
ICH |
NMPA |
|
|
高渗透性 |
药物吸收程度≥85% |
物质平衡或绝对生物利用度、人体吸收程度不少于85% |
同FDA |
绝对生物利用度≥85%或≥85%的给药剂量在尿中以原型药物,或以原型药物、Ⅰ相氧化和Ⅱ相结合代谢物的总和回收 |
同FDA |
|
|
测定 |
人体药动学研究
|
优选人体药动学测定1. 质量平衡研究2. 绝对生物利用度 |
若能证明研究设计恰当,也可接受公开发表文献中的物质平衡或绝对生物利用度数据 |
同FDA
|
可接受来自发表文献中的人体数据 |
同FDA |
|
肠渗透性 |
人体体内肠道灌注、合适的动物模型、体内或离体人或动物原肠道灌注、单层人工培养上皮细胞的离体渗透性研究 |
若采用人体肠灌流实验,应使用吸收比例不少于85%的参照化合物及阴性对照物进行方法学验证 |
证明溶液剂和固体剂型的生物等效采用标准模型药进行论证的体外渗透性研究结果 |
同FDA方法 |
同FDA |
|
|
胃肠道稳定性 |
证明药物在胃液和肠液的37 ℃孵育的稳定性,药物分解(>5%)可能代表着潜在的不稳定 |
— |
— |
证明药物在胃液和肠液的37 ℃孵育的稳定性,药物分解(>10%)可能代表着潜在的不稳定 |
同FDA |
|
目前国内对高渗透性的要求和FDA对高渗透性药物的要求一致,要求药物的吸收程度要≥85%。研究的方法主要有人体药动学研究和肠渗透性研究。其中肠渗透性研究包括人体体内肠道灌注、合适的动物模型、体内或离体人或动物原肠道灌注、单层人工培养上皮细胞的离体渗透性研究。
在仿制药的开发中,对于药物的渗透性数据,首先要查阅该产品的人体药代动力学研究数据,若说明书、审评报告或其他文献显示该产品的生物利用度≥85%或在尿液中代谢的原型药物及1相氧化和2相结合代谢产物总计≥85%,则直接说明了该产品具有高渗透性。
若无直接的人体药代动力学研究数据,则需要查阅相关文献,如关于该品种在离体动物模型的体内或原位肠道灌注的吸收程度、在Caco-2细胞中的转运情况等,若吸收程度≥85%,也可证明该药物具有高渗透性。
若上述两类数据均未查询到,则需要企业对该药物的渗透性进行科学的研究和分析,考虑到成本因素,应用较多的是体外Caco-2细胞中的渗透性研究,需要注意的是Caco-2细胞的低表达外排和摄取转运体特性,因此该方法仅适用于被动转运药物。
2.2生物等效性豁免的具体要求
普通口服固体速释制剂中的BCSⅠ和BCSⅢ类药物的生物等效性豁免条件为自制制剂与参比制剂需达到处方相似、体外溶出相似。
对于BCSⅠ类药物,只要处方中不大量使用可能影响吸收的辅料(如糖醇类:甘露醇、山梨醇、木糖醇,表面活性剂类:十二烷基硫酸钠、聚山梨酯,聚乙二醇类:PEG400等,卡波姆等),其它辅料均为国家局批准的常释制剂常用辅料,且用量与辅料在处方中对应的功能保持一致,一般不会对药物吸收速率和吸收程度产生影响。
因此,对于BCSⅠ类药物的处方要求如下:
1、可能影响吸收的辅料,种类相同,用量相似(与参比的差异在±10%以内)。
2、处方中可能影响吸收的辅料的总差异应≤10%(相对片芯)。
3、对于不影响吸收的辅料种类和用量可存在差异。
对于BCSⅢ类药物,考虑辅料可能对低渗透性药物的吸收影响更显著。因此,仿制制剂的辅料种类必须与参比制剂完全相同,影响吸收的辅料和其他辅料的用量均应与参比制剂相似(与参比的差异在±10%以内),且处方中所有辅料(含对吸收无影响的辅料)总差异≤10%(相对片芯)。法规对BCSⅢ类药物的辅料用量要求更为严格,具体见下表。
表4 BCSⅢ类药物的辅料用量要求
对于用量相似而言,对含有BCSⅢ类药物的制剂的辅料差异不应该超出以下目标值
辅料类型
参比制剂中辅料用量的百分比
可能影响吸收的辅料:
每种辅料:
差异总和:
10%
10%
相对芯部重量的百分比差异(w/w)
所有辅料:
填充剂
崩解剂
淀粉
其他
粘合剂
润滑剂
硬脂酸盐
其他
助流剂
滑石粉
其他
10%
6%
2%
1%
0.5%
2%
2%
0.2%
所有辅料(含可能影响吸收的辅料)允许的差异百分比总和
10%
在确保BCSⅠ和BCSⅢ类药物的处方满足要求以后,研发人员需经过处方工艺研究,使仿制制剂和参比制剂达到体外溶出曲线相似,提出的生物等效性豁免才有可能被允许。国法规对溶出曲线的具体要求见下表。
表5 各国法规对BCSⅠ和BCSⅢ类药物溶曲要求
|
项目 |
FDA |
WHO |
EMA |
ICH |
NMPA |
|
转速 |
篮法:100 r/min桨法:50 r/min(或者证明合适的情况下75 r/min) |
篮法:100 r/min桨法:75 r/min |
篮法:100 r/min桨法:50 r/min |
篮法:100 r/min桨法:50 r/min若经科学论证,可考虑用其他方法(例如使用沉降篮或其他恰当方法)解决堆积效应等问题 |
桨法:50/75 r/min 篮法:100r/min |
|
介质体积 |
≤500 mL(或者证明合适的情况下使用900 mL) |
≤900 mL |
≤900 mL |
≤900 mL |
≤500 mL |
|
溶出介质 |
0.1mol/LHCl或药典标准无酶人工胃液、pH4.5缓冲液、pH6.8缓冲液或药典标准无酶人工肠液。有明胶包衣的胶囊和片剂,则可以采用有酶的USP人工肠液和胃液 |
pH 1.2 的缓冲液、pH 4.5 的缓冲液、pH 6.8 的缓冲液 |
pH 1.0~1.2 的HCl 或无酶人工胃液、pH 4.5 缓冲液、pH6.8缓冲液或无酶人工肠液。推荐使用EP缓冲液对胶囊剂或用明胶包衣的片剂,可用含酶的人工胃肠液 |
3种缓冲液:pH 1.2缓冲液、pH4.5 缓冲液、pH6.8缓冲液。对于已经证明交联的明胶胶囊或明胶包衣片剂,如果得到适当证明,可能可以接受酶的使用。 |
0.1 mol/LHCL或是不含酶的模拟胃液、pH4.5 缓冲液、pH6.8缓冲液 |
|
取样点 |
应有足够的时间点,如5、10、15、20、30min |
应有足够的时间点,如10、15、20、30、45、60 min |
应有足够的时间点,如:10、15、20、30、45 min |
未作规定 |
同FDA |
2.3其他的特殊豁免类型
生物等效性豁免的两大类型为基于BCS分类的和基于多规格的豁免,其中基于多规格的生物等效性豁免,药企的实践经验相对较多,本文不作赘述。本文仅讨论其他一些特殊豁免类型。
豁免清单中的品种
对于在《可豁免或简化人体生物等效性(BE)试验品种》和《可豁免或简化人体生物等效性(BE)试验品种(第二批)》的品种,在2019年7月前可直接申请生物等效性豁免。但在2019年7月国家局发布了“部分新注册分类化学仿制药可参照《人体生物等效性豁免指导原则》进行研发与技术审评的通知”,文中明确规定豁免清单中的所有品种均需要按照《人体生物等效性试验豁免指导原则》开展相关研究,经评估符合要求的,才有可能被豁免。
因此,目前即使是开发豁免清单中的品种,也必须按照基于BCS分类或基于多规格的生物等效性豁免执行,申请人注意一定要提供充分的数据。
口服溶液剂
对于口服溶液、糖浆等溶液剂型,如果不含可能显著影响药物吸收或生物利用度的辅料,原则上可以豁免人体生物等效性试验。如果加入影响吸收的辅料,如山梨醇、木糖醇、PEG等,则可能需要做BE。可参考FDA具体品种的指南。
但目前的法规环境下,若某口服溶液在国内首次申报(首家),申请人需要结合原研药品的临床情况,评估是否需要进行生物等效性研究。
口服混悬剂
口服混悬剂通常需进行生物等效性研究。其生物等效性研究的技术要求与口服固体制剂相同。
咀嚼片
咀嚼片生物等效性研究的给药方法应参照说明书。如说明书中要求吞咽之前先咀嚼,则进行生物等效性研究时,受试者需咀嚼后吞咽给药。如说明书中说明该药可以咀嚼也可以整片吞服,则生物等效性研究时,要求以240mL水整片送服。
口服给药发挥局部作用的药物
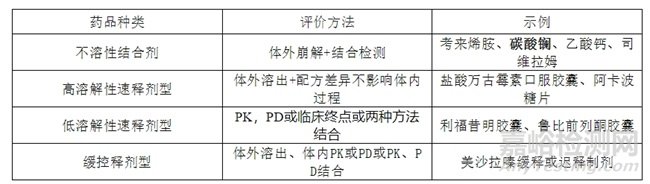
对于在胃肠道内发挥作用的药物,需根据药物特性,选用药代动力学研究、药效动力学研究或临床研究评价生物等效性,甚至可用适当的体外研究作为补充或替代评价方法。
这类情况相对复杂,一般是根据药物的溶解性进行风险评估,选择不同的研究方法。以下是几个典型的案例,如碳酸镧咀嚼片、碳酸镧颗粒剂仅需进行体外溶出研究和体外磷酸盐结合实验。
特殊的注射剂
对于特殊注射剂,由于制剂特性的复杂性,应基于制剂特性和产品特征,采取逐步递进的对比研究策略,通常首先开展受试制剂与参比制剂药学及非临床的比较研究,然后进行人体生物等效性研究,必要时开展进一步的临床研究。
《M13A: 口服固体速释制剂的生物等效性》实施建议
2024年9月国家药品监督管理局药品审评中心发布了ICH《M13A: 口服固体速释制剂的生物等效性》实施建议的征求意见稿,其中对于非高风险的口服速释制剂,法规明确可进行单个空腹条件下的研究以证明生物等效性。
美国在2024年9月13号已经率公布了根据M13A对仿制药的PSG修订计划,10月份会更新PSG,删除餐后BE要求,共计826个品种。申请人可以先关注FDA的法规动向!中国作为ICH的成员国,目前的各类药品法规紧跟ICH和FDA的步伐,预计近期可能会有针对低风险口服固体速释制剂可以免除餐后生物等效性的配套法规出现。

来源:药知晓


