您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-10-20 11:54
摘 要
Abstract
目的:为我国药典标准体系的完善提出合理化建议,增强我国药典标准组成的系统性、内容的先进性、匹配的协调性。方法:以中国、美国、欧洲、日本、英国以及国际药典标准体系为研究对象,综合采用文献法、类型比较法、深度访谈法,对《中国药典》《美国药典- 国家处方集》《欧洲药典》《日本药局方》《英国药典》《国际药典》的标准体系进行比较和分析。结果:随着科技的飞速发展,以及为加深使用者对监管目标的理解,各国及地区的药典标准体系均在体系架构、新技术标准制定、标准协调等方面不断完善。结论:我国药典标准体系应进一步完善体系架构,明确执行方式,增强与我国药品监管部门发布的指导原则的制定机制和收载内容的一致性和支持性,加强新技术标准的制定和标准的国际协调,加大鼓励企业参与药品标准工作的举措力度。
Objective: This study aims to provide rational suggestions for improving the standard system of Chinese Pharmacopoeia, enhancing the systematization of its standard composition, the progressiveness of its content, and the coordination of its alignment with international standards. Methods: This study focuses on the standard systems of pharmacopoeia in Chinese, United States, European, Japanese, British and International Pharmacopoeias. Literature review, type comparison, and in-depth interviews were used to compare and analyze the standard systems of Chinese Pharmacopoeia, the current edition of US Pharmacopoeia / National Formulary, European Pharmacopoeia, Japanese Pharmacopoeia, British Pharmacopoeia, and International Pharmacopoeia. Results: With the rapid development of technology and in order to deepen users' understanding of regulatory objectives, the pharmacopoeia standard systems in different countries (regions) are constantly improving, particularly in system framework, the development of new technology standards, and standard harmonization. Conclusion: The standard system of Chinese Pharmacopoeia should further improve its system framework,clarify its implementation methods, enhance the consistency and support of the development mechanism and content of the guidelines issued by China's drug regulatory bodies, strengthen the formulation of new technology standards, enhance international standard coordination, and increase measures to encourage enterprises to participate in drug standardization efforts.
关键词
Key words
药典;药品标准;体系;比较
pharmacopoeia; drug standard; system; comparison
标准体系是指一定范围内的标准按其内在联系形成的科学的有机整体。标准体系既是用于对标准整体进行研究、管理的模型和工具,也能够为标准制修订和应用提供科学指导,为标准资源库的建设提供资源分类关系,为各专业提供标准化对象及其要素的关联脉络。标准体系是管理因素和技术因素综合作用的产物[1]。药品标准体系的完善程度对药品安全的保障起到举足轻重的作用。药典是药品标准体系的核心,是一个国家或地区记载药品标准、规格的法典,一般由官方机构或由该国或地区法律授权的组织负责制定[2-3]。近年来,随着科技的飞速发展,以及为加深使用者对监管目标的理解,许多国家和地区的药典标准体系均在不断完善。本研究以2020 年版《中国药典》、《美国药典- 国家处方集》(United States Pharmacopoeia/National Formulary,USP/NF)现行版、《欧洲药典》(European Pharmacopoeia, EP ) 11.0版、《日本药局方》(Japanese Pharmacopoeia,JP)第18 版、《英国药典》(British Pharmacopoeia,BP)2023 版、《国际药典》(International Pharmacopoeia,IP)第11 版标准体系为研究对象,通过对上述各国及地区药典标准体系的比较研究[4],结合我国国情,为我国药典标准体系的完善提出合理化建议,增强我国药典标准组成的系统性、内容的先进性、匹配的协调性,以期推动我国药品质量控制范围从终产品检验扩大至全生命周期,使药品标准从地域性走向全球化国际协调。此外,本研究对国际各药典机构鼓励企业参与药品标准工作的措施进行了介绍,提出了我国药品标准管理机构鼓励企业参与药品标准工作的建议措施。
1、 各国及地区药典标准体系
1.1 药典标准体系总体架构
各国及地区的药典标准体系总体架构类似。目前,只有《中国药典》和BP 仍分卷册收载药品标准。《中国药典》突出各论标准,各论标准约占一级结构的87.5%。USP/NF 将启动文章、试剂和参考表格、其他资源均作为一级结构,体现了USP/NF 内容的多元化。EP 将包装容器和材料通则、试剂、产品总论、制剂通则均作为一级结构,强调了对不同类型产品的控制要求。JP 将植物药通则、红外光谱图和紫外光谱图作为一级结构,体现了对于植物药标准和配套标准的重视。BP 的一级结构以各论标准为主,与其他药典不同的是,BP 将药用物质(即原料药和辅料)与制剂单独收载,将红外光谱图也作为一级结构。IP 将凡例的辅助说明、红外光谱图、试剂试药等作为一级结构,更好地帮助使用者执行相关要求(图1~6)。
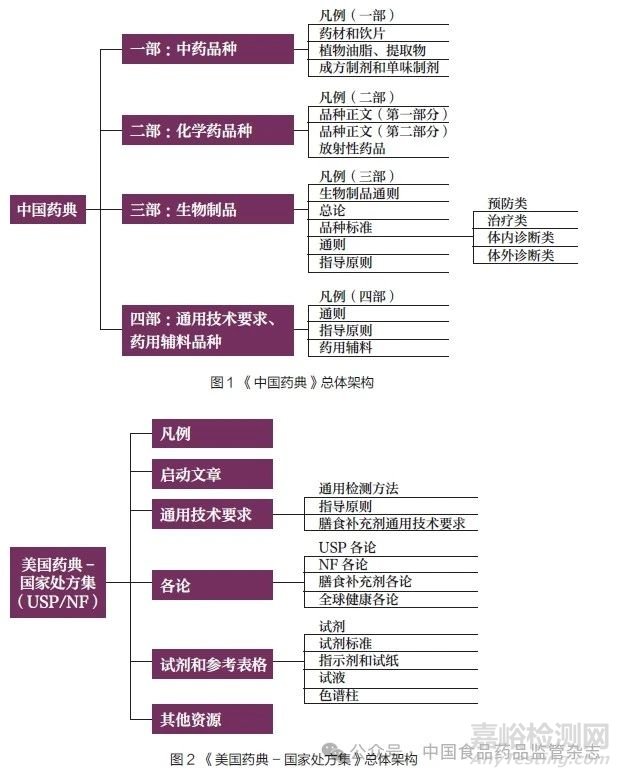

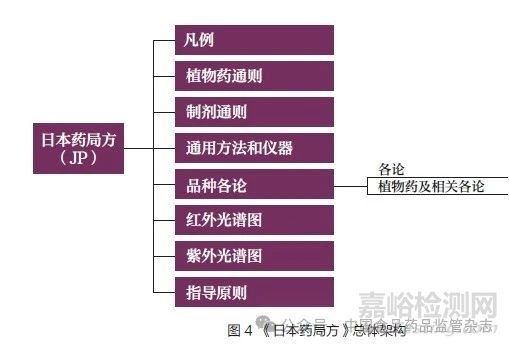


从收载范围上看,由于各国及地区药品监管法规存在差异,以及受药典外标准的影响,除药品标准外,USP/NF 收载了膳食补充剂标准;EP 收载了顺势疗法制剂标准;BP 收载了外科手术用材料、兽药标准。从标准配套信息收载上看,JP、BP、IP 均收载了部分药品的红外光谱图或紫外光谱图;USP/NF 提供了有关色谱柱信息等更多资源。从标准类型收载上看,USP/NF 收载了新型标准“启动文章”,阐述新的、前沿的标准制定意向或计划,提前介绍下一步标准增修订工作,以便利益相关方提前知晓和参与。
从收载数量上看,不同专业领域的各类型标准的数量不同。《中国药典》收载中药各论标准最多。除《中国药典》外,我国还存在其他局(部)颁等标品种标准。USP/NF 收载的化学药原料药、辅料和制剂各论标准最多,但收载的生物制品标准较少,这是由于美国《联邦法规汇编》(Code of Federal Regulations,CFR)第21 篇“ 食品与药品”(Title 21: Food and Drugs)600~660部(Part 600~660),以及美国食品药品监督管理局(Food andDrug Administration,FDA)发布的相关指南涵盖了生物制品的质量控制和生产。2010 年以前,USP/NF 收载了比现在更多的生物制品品种标准,但在2010年左右被删除,原因包括:缺乏质量信息,不再在美国销售,只提到了检测项目但没有提供方法,许可证被吊销,不再以同样的方式生产,或者生产被纳入FDA 关于生物制品的法规范畴等。USP/NF收载的通用技术要求标准最多。EP 只收载了少量制剂标准,制剂产品的质量需要符合各成员国药典或药品管理当局批准的质量标准要求。JP 收载的各类型标准均较少,这是因为日本还存在大量JP 外的法定标准,如日本生物制品最低要求、日本放射性药品标准、日本药局方外标准、日本药局方外生药标准、日本药用辅料标准等。根据BP 与欧洲药品质量管理局(European Directorate for the Quality of Medicines & Healthcare,EDQM) 签订的合作协议,BP复制了EP 的标准,还收载了BP特有的标准。IP 主要收载化学药品和辅料标准,对于放射性药品的通用技术要求收载较多。
1.2 药典各类型标准执行方式
各药典均在凡例中规定了各类型标准间的关系和执行方式。各药典中的各类型标准都不能单独应用,应结合凡例、总论、通则、各论来执行。USP/NF、EP和BP 均允许不必执行各论标准中的所有测试,具体分为两种情况:一是在证明替代方法和药典方法等效的情况下,可使用经主管当局批准的替代方法;二是在不需要证明替代方法和药典方法等效的情况下,药典各论中明确允许选择不同的检测方法以反映不同生产商的品种特性。
EP 对于各类标准的执行要求较为严谨,主要体现在以下4 个方面:一是与其他药典不同,EP认为只有在品种标准引用时,通则及通则引用的内容才具有强制性,否则仅供参考。二是EP 中收载的大量总论标准是其特色。EP 明确了总论适用于所定义类别的所有产品。尽管各论标准一般不引用总论标准,但均应在执行各论标准的同时执行相应的总论标准。三是明确制剂通则适用于所定义剂型的所有药品,无论其是否为药典品种。对于特定的药品,制剂通则的要求不一定全面,主管当局可能会对制剂通则中的规定提出额外的要求。四是与“除另有规定外”(unless otherwise specified)一词(表示当存在与凡例或通则有关规定不一致的情况时,在各论标准中另作规定,并按该规定执行)相区别,考虑到主管当局批准的特例情况,明确了“除另有正当理由和授权”(unless otherwise justified andauthorised)、“适宜”(suitable/appropriate)的定义。
1.3 药典草药标准体系
1.3.1 数量和类型
USP/NF、EP、JP 均收载草药300 余种,《中国药典》收载的中药各论标准数量远多于其他药典。《中国药典》和USP/NF收载的草药制剂标准占其草药标准的最大比例。《中国药典》收载的中药制剂各论标准远多于其他药典。EP、JP 收载的药材标准占其草药标准的最大比例。2014年,原国家食品药品监督管理总局发布《关于加强中药生产中提取和提取物监督管理的通知》,要求对中成药国家药品标准处方项下载明且具有单独国家药品标准的中药提取物实施备案管理。对属于备案管理的中药提取物,可自行提取,也可购买使用;对不属于备案管理的中药提取物,应自行提取。依据此规定,提取物是否具有单独的国家药品标准,会对其监管方式产生影响。综上,鉴于我国植物提取物的监管形式、单独的国家标准总数较少等因素,《中国药典》收载的植物油脂和提取物、植物原料的数量较USP/NF和EP 少。
1.3.2 项目设置
以上多数药典均对草药建立了相应的质量标准,设置的项目也大体相同,包括来源、鉴定、检查、含量测定等。与其他药典相比,《中国药典》还设置了医学信息相关项目,如用法与用量、功能与主治、注意项目以及炮制相关要求。《中国药典》与JP 均收载了制法项,但两者均未收载放射性物质残留控制项目。JP 未收载指纹图谱、特征图谱化学鉴别以及贮藏项目。只有USP/NF 设置了标签和对照品项目。上述药典的具体检测标准的差异主要体现在安全性控制方面,集中体现在农药残留限量、重金属和有害元素限量以及微生物限量这3 个指标。
1.4 药典化学药品各论标准体系
1.4.1 数量和类型
USP/NF 收载的化学药品各论标准总数远高于其他国家和地区的药典。EP 收载的制剂标准数量较少,仅占3%。JP 收载的原料药各论标准较多。《中国药典》、USP/NF、BP 收载的制剂标准数量大于原料药标准数量。IP 收载的化学药品各论标准总数较少。
1.4.2 项目设置
通过研究各药典共有的化学药品各论标准发现,在质量控制项目数量方面,《中国药典》及EP、JP 的质量控制项目设置较多,USP/NF、IP 的质量控制项目设置较少。对于USP/NF,因部分标准检测项目设置和检测方法较为先进,故项目设置较少。IP 聚焦于质量控制中最基础的方面,适用于不同来源的样品,对个体特定的检测项目未作规定,故项目设置较少。各药典均有各自独有的质量控制项目,如《中国药典》的降压物质、引湿性等。对于有关物质,USP/NF、EP 控制较严格,设置的质量控制项目较多,方法更加先进,而IP 设置的检测项目较少。BP、IP 均指出,各论中的杂质分析方法旨在指导分析需要关注的杂质,不能认为不需要控制未列明的杂质,应根据产品的处方和工艺对其可能存在的杂质进行合理控制。除《中国药典》外,其他药典均执行各自收载的基于国际人用药品注册技术协调会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)Q3D 指导原则的元素杂质通用技术要求。EP、JP部分各论标准中,有“生产”质量控制项目。JP 将各论中“生产”中的内容定义为提请企业注意生产过程的特定方面,但不一定详尽无遗。除另有规定外,为强制性要求。这些要求不一定由分析人员在终产品上进行验证。主管当局可以通过检查从企业收到的数据、检查或测试样品来确定企业是否遵循相关规定。需要说明的是,标准中没有“生产”部分并不意味着不需要注意上述特征。
在限度指标方面,不同药典在不同项目的限度宽严各不相同。
在各论检测方法方面,因考虑到发展中国家的需求,IP 采用的检测方法较为经典简便,但在灵敏度、准确度方面不如其他药典。在部分各论标准中,IP 给出了两种方法,针对具备不同检测能力的机构设定可选择的方法。USP/NF 在部分各论中允许选择不同的检测方法以反映不同企业的品种属性,如不同的晶型、杂质、水合物和溶出度。EP 指出,如果某一药品不符合各论标准溶出度试验要求,但该产品获得了监管部门的批准,则监管部门应提请欧洲药典委员会注意,以便其在审查各论标准时进行修订。此外,对于一些先进的方法, 如过程分析技术(process analytical technology,PAT)、实时放行检测(real time release testing,RTRT)可以作为终产品检测的替代方法。在监管部门认为适当的情况下,可以采用RTRT,并不需要因遵守EP 相关规定而将其排除。
1.5 药典生物制品各论标准体系
1.5.1 数量和类型
《中国药典》生物制品各论标准收载总数略多于其他药典,标准体系整体全面。各药典生物制品标准均以治疗类生物制品为主要类型,均占半数以上,收载数量由多至少分别为《中国药典》、JP 和日本生物制品最低要求、EP、USP/NF。治疗类中的主要分类有胰岛素类、免疫球蛋白类、干扰素类,在各药典标准中皆有出现,但数量占比不尽相同。其次是预防类生物制品,收载数量由多至少分别为EP、《中国药典》、USP/NF、JP 和日本生物制品最低要求。
因法律框架关注不同,各药典生物制品标准收载类型和数量不同。例如, 生物制品体外诊断产品为《中国药典》所特有。而在EP 中,体外诊断产品受体外诊断器械指令监管,不受药品法规监管,其高风险类别产品的质量标准在欧洲通用规范(Common Specification,CS)中进行了描述,体外诊断产品超出了EP 收载范围。在美国,体外诊断产品由FDA 医疗器械与放射健康中心(Center for Device and Radiological Health ,CDRH)监管。高风险体外诊断产品的质量标准要求可在特定产品的指导原则中体现,产品登记必须通过上市前批准途径提交,或至少应与市场上现有产品等同,产品登记可能需要通过510(k) 或相关途径提交。在日本,体外诊断产品也不在其药典的收载范围。至于体内诊断产品,只在EP 与《中国药典》收载了相关标准,占生物制品的极小部分,EP 为6.6%,《中国药典》为2.6%。
1.5.2 项目设置
与其他类型制品不同,各药典生物制品各论标准均设置了生产过程项目,规范了需要控制的各阶段性产品,亦引入了与过程依赖性异质性相关的测试。各药典对于不同阶段产品的项目设置、检测方法各不相同。《中国药典》原液和制剂部分均包含鉴别、纯度、含量测定、微生物检查等安全性和有效性方面的重要项目。在部分项目中,USP/NF、EP 没有检测方法要求,只有接受标准要求,或规定可使用经监管机构批准的方法和限度。相比于其他药典,JP 对终产品前的各阶段产品的质控项目较少。《中国药典》对每个检测项目的检测方法均为药典特定唯一方法,与USP/NF、EP、JP 相比, 各论的检验项目总体有更为明显的强制性。USP/NF 和EP 中大部分检测项目没有规定药典特定方法,可灵活选择使用非药典收录的多种方法。
1.6 药典药用辅料标准体系
USP/NF 收载的药用辅料各论标准多于其他药典,JP 收载的药用辅料各论标准较少。在标准体系的架构上,《中国药典》、USP/NF、EP、JP 均建立了以凡例、各论、通则、指导原则为支撑的药典药用辅料标准体系。各药典均在凡例、各论、通则、指导原则中提供了药用辅料的定义、标准执行方式、药用辅料性能和安全性评价指南。从各论标准的收载方式上看,《中国药典》和USP/NF 均将药用辅料各论标准单独分类收载,而其他药典将药用辅料各论标准与其他各论标准混编。USP/NF 建立了一系列与药用辅料生命周期有关的指导原则,如药用辅料的药品供应和管理规范(Good Distribution Practice,GDP)、药品生产质量管理规范(Good Manufacturing Practice,GMP)、分析证书(certificate of analysis,COA)、变更管理指导原则、供应商审计指导原则等。EP 在药用辅料各论标准中附加了药用辅料功能性指标(functionality-related characteristics,FRCs)列表,还给出了一种或多种测定这些特性的分析方法以供参考。药品制造商有责任根据药用辅料使用和药品研发数据,决定如何在制造过程中应用FRCs 信息。JP 明确指出,当JP 收载了药用辅料标准,则该辅料要满足标准中的限度。USP/NF 也给出了药用辅料各论标准适用性和合规性的规定。
1.7 药典药包材标准体系
各药典均以通用技术要求形式收载药包材标准,并共同关注了药包材材料的质量以及药包材的适用性、相容性与安全性。各药典的体系架构有所不同,《中国药典》以收载通用检测方法为主,USP/NF 多收载涉及不同种类药包材的使用评价、完整性评价、功能性评价、相容性研究评价等的指导原则。EP 收载了较多容器生产用材料通则。JP 则在制剂通则、通用检测方法和指导原则中分别收载药包材总体要求、试验方法、基本要求和术语以及评价方法等内容。
1.8 药典通用技术要求标准体系
1.8.1 数量和类型
按照药品全生命周期各环节17 个维度的分类,即研发、命名、术语、原料和辅料、生产、制剂、分析方法、分析方法验证、标准物质、实验室质量管理、仪器设备、试剂、包材、标签、运输和贮存、使用、国际协调。USP/NF覆盖其中的15 项,《中国药典》覆盖14 项,EP、JP 各覆盖12项,BP 覆盖10 项,IP 覆盖4项。除IP 外,其余药典均涉及药品全生命周期中较多环节的要求。其中,USP/NF 覆盖率最高,近90% ;《中国药典》覆盖超过80% 的环节。各药典按照通用技术要求总数量从多到少依次为:USP/NF、EP、《中国药典》、BP、JP、IP。
除IP 外,其余5 个药典均收载了原料和辅料、分析方法验证、试剂相关的指导原则。有4 个药典收载了生产、仪器设备、包材、运输和贮存相关的通用技术要求。有3 个药典收载了研发、命名、术语相关的通用技术要求。有2个药典收载了实验室质量管理、标签、国际协调相关的通用技术要求。由于药典收载内容与患者并不直接相关,目前只有USP/NF收载了与使用有关的通用技术要求。
各药典在制剂、分析方法、标准物质方面均制定了通用技术要求。其中, 分析方法通用技术要求占比均为最高, 均超过75%。尽管ICH Q4 指导原则和药典讨论组(Pharmacopeial Discussion Group,PDG)协调案正在实施和发展,但各药典在部分通用的分析检测方法、局部应用产品(如吸入、鼻、眼用产品)和皮肤给药产品的检测方法等方面仍存在差异。《中国药典》虽收载了部分先进的分析技术通则,但在各论标准中的应用方面仍有待加强。
在制剂通则方面,除USP/NF外,各药典收载方式相似,均明确了制剂的定义、生产、测试、包装、贮存、标签等方面要求,以及各类型亚剂型的质量控制要点。USP/NF 主要侧重于各类产品的质量控制测试方法。《中国药典》生物制品总论与EP 产品总论的收载结构类似,包含制剂通则中所描述的几大方面,以及特定产品的质量控制额外要点。与其他药典不同,EP 在部分制剂通则和产品总论的第一段明确该文本的适用、不适用和不一定适用的范围,并给出了针对不适用和额外要求参考的文本信息。EP 指出,在特定条件下,制剂通则和产品总论的要求不一定全面,补充或附加要求可以在单独的各论中规定,也可以由主管当局实施。JP 指出,制剂通则对测试方法的描述是基本要求,生产方法代表了常用方法。BP 产品总论还帮助使用者更好地了解各论制定过程和各论制定的考虑。
1.8.2 与药品监管机构发布的相近内容指导原则的比较
笔者从各药典机构和相应药品监管机构发布的相近内容指导原则(通用技术要求)的角度,比较和分析两者的制定目的、适用范围和具体内容的异同点和相互引用情况[5],发现两者的关系呈现以下特点。
一是主题分布方面,在制剂、生物制品、稳定性、杂质、溶出度、微生物检测、分析方法验证等方面,存在较多相近内容的指导原则(通用技术要求)。
二是内容侧重方面,两者在制定目的和侧重点上有所差异。药典通用技术要求侧重于各项技术、方法的具体要求;药品监管机构发布的指南重点关注注册申报的要求和研究技术。
三是冲突和重复方面,除我国外,其他国家或地区药品监管机构和药典机构发布的相近内容指导原则(通用技术要求)中重复内容较少,没有相互冲突的内容。通常情况下,两者内容相似的指导原则(通用技术要求)会引用相同的国际标准,如ICH 和ISO 标准。
四是互操作性方面,相比于监管机构发布的指南引用药典通用技术要求的情况,药典通用技术要求引用监管机构发布的指南较少。监管机构发布的指南促进了药典通用技术要求的符合性。药典通用技术要求和监管机构发布的指南之间往往相互补充,指南里若涉及具体的方法技术一般会引用药典的相关通用技术要求。两者共同支撑药品标准体系。
2、 各国及地区药典新技术标准制定趋势和药品标准协调方向
在新技术标准制定方面,各药典不断加强新技术标准的制定,以应对科技的快速发展、加深使用者对监管目标的理解,体现出标准应尽快反映技术进步成果和市场需求的原则。《中国药典》、USP/NF、EP、JP、BP 均在生物制品、杂质、连续制造、分析方法源于设计、动物试验替代方法方面布局, 制定新技术标准,这些领域也是各药典潜在的合作和竞争领域。在国际协调方面, 各国及地区均不断加强药品标准的国际协调, 积极参与世界卫生组织(World Health Organization,WHO)、ICH、PDG 等国际多边或开展双边药品标准制定活动。各药典国际协调的一般策略是:以科学研究体系为基础,完成一系列国际标准的研究及制定工作;形成国际协调共识,成立多方合作专家委员会,建立良好的国际标准制定对话机制;开展学术交流,带动标准理念的协调与共识,逐步实现主导国际药品标准的目标[6-9]。
3、 各国及地区药典机构鼓励企业参与药品标准工作措施
各药典机构通过荣誉奖励,主动寻求和邀请的方式鼓励相关企业参与标准工作,充分调动企业积极性,推进建立以企业为主体的标准工作机制。我国药品标准管理机构主要采取以下措施鼓励企业参与药品标准工作。一是建立观察员制度。从2017 年第十一届国家药典委员会组建起,设立观察员单位,邀请相关行业协会、学会代表作为观察员列席相关专业和学术会议,进一步体现药品标准工作的开放性和包容性。二是在官网发布鼓励企业参与样品征集以及标准调研、制定、研讨的通知,并公布提供样品及资料的企业名单。三是建立国家药品标准提高课题招标制度。鼓励企业承担或参与标准研究工作,增加药品标准提高的推动力。四是在发布国家药品标准或者省级中药标准公示稿时,标注药品标准起草单位、复核单位和参与单位等信息。
USP/NF 中的标准大多数为企业提出。USP/NF 对捐献标准和物料的企业奖励水晶奖杯、证书、感谢信,并每年给予捐献积分奖励(根据捐献获得相应积分,在USP/NF 相关服务, 如购买标准、标准物质、培训等中获得抵扣)。BP 鼓励企业就标准提出修订建议和相关详细信息,并指出尽早提交建议符合企业的利益。同时,BP 向企业寻求必要的信息,鼓励企业就其感兴趣的标准提供适用的验证数据。为了提高企业提交草案的质量,JP 公开了草案编制的纲领性文件。IP 鼓励企业捐赠合适的候选材料,为国际化学标准品(international chemical reference substances,ICRS)的可用性做出贡献。此外,IP邀请企业向WHO 药品资格预审小组提交产品评估的意向书(expression of interest,EOI),分享各论标准中包含的相关信息。
4、 对于完善《中国药典》标准体系的思考
4.1 完善《中国药典》标准体系
完善标准体系总体结构。增加《中国药典》通用技术要求的收载,对《中国药典》一至四部的凡例进行规范统一,增加产品总论标准的收载,对《中国药典》指导原则进行分类,并扩充指导原则的功能,将试剂标准作为一级结构收载。在凡例中进一步明确各类型标准的执行方式,当监管部门批准不同于药典标准的注册标准时,及时将其反馈至药典机构。
完善各类型药典标准体系。一是对于中药标准体系,加强整体性和安全性控制。二是对于化学药品标准体系,加大各论标准的收载数量,完善质控项目的设置,考虑替代方法的实施可能性。三是对于生物制品标准体系,加强新产品总论标准的制定,完善质控项目的设置,增加各论标准执行的灵活性,继续倡导替代、减少和优化的3R(replacement,reduction,refinement) 原则。四是对于药用辅料和药包材标准体系,增加制剂所需药用辅料各论标准的收载,加强药用辅料和药包材通用技术要求的制修订,优化药用辅料各论标准质控项目的设置,加强功能性相关指标体系的建立,增强标准的灵活性,进一步明确在我国原辅包关联审评审批制度下药用辅料和药包材各类型标准的适用性和执行方式。五是对于通用技术要求标准体系,增加产品总论的制定,增加支持药品研发、生产的分析方法通用技术要求的制定,增加药典制定程序相关的通用技术要求的制定,增加仪器设备、术语、命名、国际协调相关通用技术要求的制定。
进一步增强《中国药典》通用技术要求与我国药品监管技术指南的一致性和支持性,由药品监管部门牵头确定主题,进一步优化完善协调机制。根据各机构的职能,协调、统一药品监管部门和药典机构制定的指导原则(通用技术要求),确保两者在制定目的、适用范围和收载内容上定位清晰,并及时更新有冲突的通用技术要求。加强两者的相互引用,即互操作性,并与国际标准保持同步。此外,发挥各类型标准在我国药品监管中的合力,增强标准的执行效果。
4.2 加强新技术标准的制定和药品标准的国际协调
促进数字技术、高通量测序技术、纳米药物、复杂产品、生物制品和先进治疗产品通用技术要求或各论标准的制定,从而支持创新药物的研发。建议各论标准加大对先进分析方法的应用。将分析质量源于设计(analytical quality by design,AQbD)、分析方法生命周期理念融入药典各论的制定中,提高对产品从研发到上市后监管各阶段所面临挑战的理解。积极参与ICH Q4 指导原则的协调,探索开展对前瞻性标准的双边协调,引入依属性协调的概念,从部分内容协调开始,逐步推进药品标准的国际协调。加强与国外药典机构的交流合作,对共同感兴趣的领域进行知识共享。培养标准国际化人才,促进其他国家和地区将《中国药典》作为国际标准。
4.3 加大鼓励企业参与药品标准工作的举措力度
加强药品标准工作的宣传力度,增加来自于企业的药典委员数量,对在国家药品标准工作中表现突出的企业予以表彰奖励,将药品标准工作纳入各级奖项评选范围,并作为技术职称晋升依据。鼓励企业开展新技术标准研制工作,建立科研成果向标准转化的机制,鼓励制定团体标准,探索建立政府颁布标准采信市场自主制定标准的机制。优化标准制定流程和平台,健全企业参与标准制修订的机制。定期开展药品标准培训工作,建立药品标准人才长效培训机制。

来源:中国食品药品监管杂志


