您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-10-28 08:24
心脏的功能依赖于数百万被称为心肌细胞的杆状肌肉细胞的协调作用,这些细胞随着每次心跳而纵向收缩。如果心脏要有效地泵血,心脏每个区域的肌细胞必须在正确的时刻施加力。这种收缩的精细时间由动作电位控制,动作电位是细胞电位在几分之一秒内的周期性变化。动作电位是由离子通道蛋白的打开和关闭产生的,离子通道蛋白允许钠、钾、氯和钙离子在细胞膜上运动。
hERG通道是指人类心脏中的一种钾离子通道,由hERG基因编码,全称为human Ether-a-go-go Related Gene。这个通道也被称为Kv11.1,是钾离子通道的α亚基,对心脏的电位活性至关重要,主要负责介导心脏动作电位中的延迟整流钾电流(IKr)的复极化阶段。即在下一轮去极化之前将膜电位恢复到静息状态。通过阻断hERG通道,许多药物可以延迟复极,随着这种延迟,心肌细胞容易过早激活和收缩。心脏特定区域的过早信号会引发不规则的心跳,包括尖端扭转性室速(Torsade de Pointes, TdPs)。
QTc间期延长和TdPs是20世纪90年代停药或限制药物上市的重要原因,并导致ICH S7B和ICH E14指导原则的出台,提示药企需要重视药物对hERG或IKr通道、临床前和临床QTc的影响。有数据显示,60%处于研发阶段的药物可以抑制hERG,15%上市药物会引起QT间期延长,4%上市药物会引起TdP心律失常。不过,研究表明,除了hERG之外,许多药物还同时与其他电压门控通道相互作用,并且这些相互作用可以减轻由于hERG抑制而导致的TdP风险。
ICH S7B和ICH E14批准的近20年时间,对TdPs的机制以及早期后去极化在心律失常中的作用有了更深入的了解,也导致了综合体外致心律失常试验(Comprehensive In Vitro Proarrhythmia Assay, CiPA)的出现。CiPA侧重于评估药物的致心律失常和引起TdPs的机制。在这种方法中,即使药物抑制hERG或导致QTc延长,但如果从致心律失常的角度来看是安全的药物,将被鉴定和标记出来。
当然,本篇文章的主角是hERG,一种电压门控钾离子通道,如下图所示,hERG是由4个相同亚单位组成,每个亚单位包含6个跨膜螺旋区(S1-S6)。其中,S1-S2由24个氨基酸连接,S3-S4由4个氨基酸连接。S4包含5个带正电荷的残基,发挥电压传感作用(voltage sensor)。S5和S6共同形成通道孔。

大多数hERG抑制剂是含有碱性氨基的小分子,在细胞膜去极化诱导的通道开放时到达胞内结合位点。对hERG结构研究发现,该通道含有一个由4个疏水口袋包绕的小的内腔。腔内的高负电性可以结合大多数hERG抑制剂携带的正电铵根。而4个疏水口袋为hERG抑制剂提供了更多结合位点。
传统观点认为,多肽和蛋白药物抑制hERG的可能性很低,因为扩散透膜能力比较差,不能到达hERG孔道经典结合位点。不过,生物药物其实可以通过结合非传统非经典位点(’unconventional’ noncanonical binding sites),影响hERG离子通道功能。
多种电压门控离子通道(Kv1.2, Kv3.1, Eag1, Nav1.5, Cav2.1/2, TRPC1, TRPC5, TRPM3, TRPV1)在S5和S6之间均包括一条长的胞外区环状结构。有研究显示,这一区域正是抗体结合并抑制电流的关键表位。比如某些患者出现抗Ro抗体阳性很可能意味着患有自身免疫性疾病如系统性红斑狼疮,这类患者可能出现复极异常(QT间期延长和TdP室性心律失常),原因与抗Ro抗体结合S5-S6之间的环状区域(氨基酸574-598)有关。这段氨基酸序列与Ro-52抗原有一定同源性。不过,单抗的分子量大(50-155KDa),不易透过细胞膜,特异性强,意味着延长QTc间期风险很低。实际上,Jackson等人在QT-prolonging effects of monoclonal antibody drugs in humans: a systematic review of two literature and a public adverse event database一文中统计过欧洲批准上市的28个单克隆抗体药物,发现所有这些抗体药物均未见QTc间期延长风险。所以,对于单抗类药物,ICH E14也没有要求临床期间开展详细的QT研究。临床前通常也不需要开展hERG试验。ICH S6只是建议在慢性动物毒理研究中伴随考察心血管安全药理学指标。
雌激素(Estrogen)是一类主要的女性性激素,对女性生殖系统和第二性征的发育和维持至关重要。女性出现药物诱导的TdP心律失常风险高于男性,其中一个原因可能与雌激素抑制hERG离子通道有关。使用分子对接发现,雌激素与hERG有两个结合口袋,一个位于胞内经典结合位点-内腔,一个位于胞外孔道环状结构区域,即上图中的turret位置。
传统认为,寡核苷酸分子量大,直接作用于hERG离子通道的可能性很低,故普遍认为寡核苷酸类药物开展hERG试验的意义不大。不过,有个名为ISIS345198的反义寡核苷酸,在75μM及以上浓度会抑制hERG。而这个分子的分子量>5KDa,并具备高电荷、亲水性特点,不太可能穿过细胞膜,无法结合在胞内经典位点,很大可能也是结合在胞外区发挥的作用。一款靶向双调蛋白(amphiregulin)的siRNA(SAMiRNA-AREG)在浓度≥20μg/mL时,会抑制hERG通道。心脏主要miRNA(miR1)通过结合K离子通道Kir2.1胞内区C端,可以抑制K离子内流。实际上,为排除这一风险,大部分已上市的寡核苷酸类药物均开展了体外hERG试验。比如治疗家族性高胆固醇血症的mipomersen、用于造血干细胞移植后的肝VOD的defitelio、治疗遗传性转甲状腺素蛋白淀粉样变性的inotersen和patisiran等。
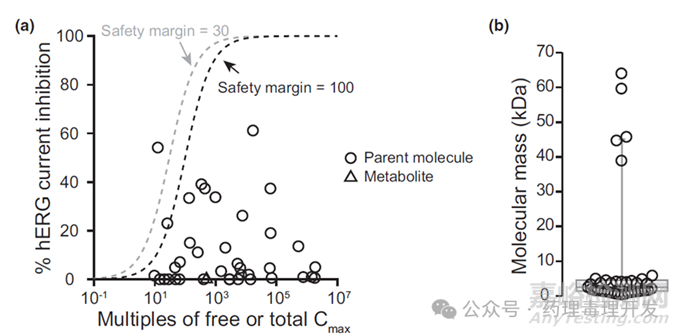
那么多肽或者分子量比抗体小的蛋白对hERG的影响如何呢。Wu等人研究过在FDA申报的38种多肽和蛋白对hERG的影响,并以题为ICH S7B in vitro assays do not address mechanisms of QTC prolongation for peptides and proteins-data in support of not needing dedicated QTC studies的文章发表。发现在高浓度下,2个产品出现≥50% hERG抑制,8个产品出现20-40%抑制。不过,未见体内QTc间期的延长,可能与临床暴露浓度低于体外hERG抑制浓度有关。如下图所示,左图显示的是hERG抑制与临床Cmax之间的关系,38个产品中,采用100倍或者30倍的安全窗口,≥95%的产品hERG抑制均是阴性。右图显示的是38个产品的分子量分布,其中33个(87%)产品的分子量低于6KDa。
最后
很明显,大分子对hERG通道的抑制风险要低很多。但即便如此,考虑到心脏安全性几乎是新药开发中的一票否决项。很多多肽类药物还是进行了hERG研究,比如已上市的GLP-1受体激动剂利拉鲁肽、度拉糖肽、利司那肽、司美格鲁肽、替尔泊肽等。毕竟回顾性分析,少量多肽类药物确实存在抑制hERG风险,需要根据hERG抑制浓度和临床拟用剂量下的暴露浓度评估安全窗口。寡核苷酸类药物的分子量也很大,并具备高电荷、亲水性特点,不太可能穿过细胞膜,理论上作用于hERG的风险较低,但实际上已上市的寡核苷酸类药物也大都进行了hERG研究,如前文所述,并不能排除hERG抑制风险。分子量较大的单克隆抗体因特异性和透膜能力原因,hERG不做要求,如果在体内试验中发现心脏安全性风险,可以回头研究是否是影响了hERG通道所致,正如文中抗Ro抗体案例所示。

来源:药理毒理开发


