您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-11-05 18:47
本文使用一组已通过生物等效性正式试验的高变异药物研究数据,进行上百次模拟生物等效性预试验,统计主要药代动力学参数(Cmax、AUC)的几何平均值和个体内变异系数的概率,据此推测高变异药物生物等效性预试验通过的概率。结果显示,主要药代动力学参数的几何均值具有较大的随机性和不确定性,个体内变异系数受个体受试者数据影响较大。采用两周期交叉试验设计开展生物等效性预试验不能有效地指导高变异药物制剂处方工艺开发及生物等效性正式试验。
随着仿制药质量和疗效一致性评价工作的深入开展,药物研发机构一般会选择采用两周期交叉试验设计开展生物等效性预试验研究,为制剂处方工艺开发及正式生物等效性试验提供数据支持。对于 BCS1 类或 3 类药物,开展生物等效性预试验,一定程度上加快了药物研发进程。而对于高变异药物(指某些药物由于生物利用度过低、酸不稳定、吸收前的广泛代谢等原因,导致一个或多个药动学参数的个体内变异系数(Within-subject Coefficient of Variation,CVW%)大于或等于 30%。采用两周期交叉试验设计法获得的生物等效性预试验数据是否能够有效指导制剂处方工艺开发及正式生物等效性试验,业内存在较大争议,主要观点如下:
(1)为生物等效性试验样本量估算提供依据:国家药品监督管理局(NMPA)、美国食品药品监督管理局(FDA)[1] 相关指导原则均建议正式试验开始之前,可在少数志愿者中进行预试验。预试验可用于验证分析方法、评价变异情况、优化采样时间,并获得其他相关信息。用于评价变异情况的药代动力学参数(药峰浓度 Cmax 和药物曲线面积 AUC)几何均值比以及个体内变异系数(CVW%)等,并据此进行研究方案设计,计算合适的受试者例数。
(2)为制剂处方工艺开发参考依据:如没有建立体内外相关性较强的溶出曲线评价方法的话,可采用生物等效性预试验评估仿制制剂与参比制剂处方工艺的差异,主要药代动力学参数(Cmax 和 AUC)几何均值比以及个体内变异系数(CVW%)如与参比制剂有明显差异,为制剂处方工艺优化调整提供方向[2]。
(3)完全没有必要开展生物等效性预试验:对于高变异药物,如采用常规的受试者例数(一般为 12 例),个别样本变异较大,会影响生物等效试验参数的计算结果,有时即使参比制剂与自身相比较,也可能出现不能证明其生物等效的情况[3],生物等效性预试验结果会出现假阳性或假阴性数据。
高变异药物生物等效试验的样本量对于药物的生物等效试验非常重要,由于药物在个体内的变异大,数据的离散程度也就比较大,使用常规样本来进行生物等效试验很可能会把生物等效的药物误判为不等效的药物,样本量过大又涉及到成本和伦理方面的问题,因此对高变异药物的生物等效的样本估算是非常重要的,高变异药物生物等效试验样本量估算产生最大影响的主要是参比制剂的 CV 值和受试制剂与参比制剂之间的几何均值比。
我们参考 Isabel Moreno 等人证明最有效的 12 个受试者单次给药双交叉的模拟预试验设计,按照预试验的结果来分别计算对正式试验入组例数估算最重要的两个因素变异系数 CV 值和几何均值[4]。尤其通过调查是同一个参比制剂在预试验中的 CV 值和几何均值的分布范围,进而了解如果参比制剂和受试制剂如果在药学上非常接近,预试验是否能很好地预测正式试验的结果和正式试验需要的受试者例数。同时,也考察对于AUC的变异也超出30%的情形下,预试验是否能够很好地预测 AUC 的比值。
有很多专家学者认为预试验结果中,数据的“单向偏移”(大多数受试者的两次暴露量的比值均大于或小于 1)可以用来定性的预测受试制剂和参比制剂的暴露量的关系,并应用这个结果来决定是否开展正式生物等效试验或者用来指导制剂的进一步开发。我们对这样的一个假设表示怀疑,我们也利用这些数据来模拟计算这样的假设是否正确。
1、试验方法
生物等效性试验数据:某高变异药物随机、开放、三周期、部分重复健康成人生物等效性试验数据,受试者例数42 例。
模拟试验:采用 DAS 2.0 软件对生物等效性数据进行计算药代动力学参数,并进行等效性评价。参考 IsabelMoreno 等人过去采用的方法[4],在一个已经通过生物等效的高变异药物的试验数据中,按照 2×12 的取样方法来进行模拟的预试验,采用 Excel 软件中的函数取随机号,分别抽取 100 次,每次都计算每一次模拟预试验取得数据的几何均值和 CV 值。然后对这 100 次结果的几何均值和 CV 值的分布进行统计。
2、试验结果
2.1 用于生物等效性试验样本量估计的风险分析
采用两周期交叉试验设计法对同一批参比制剂进行生物等效性试验研究,在 N=42 的条件下,主要药代动力学参数 Cmax、AUC(0-t)、AUC(0- ∞)几何均值比值的 90% 置信区间数值符合要求;按照随机号顺序分为 2 组,即N=21 的条件下,出现了第一组(1 ~ 21号)等效,第二组(22 ~ 42)不等效的情形;按照随机号顺序分为 2 组,即N=14 的条件下,三组结果均不等效。因此,采用两周期交叉试验进行生物等效性预试验研究获得体内变异系数进行正式生物等效性样本量估计以及临床研究方案设计,存在较大的风险。
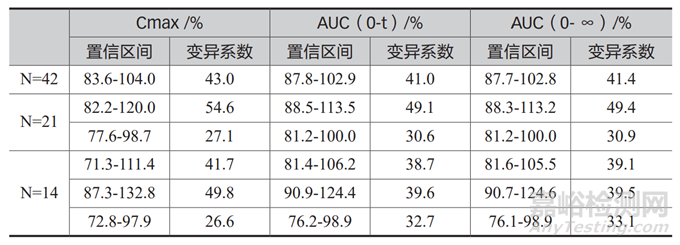
表1 不同样本量条件下参比制剂药代动力学参数体内变异系数及90%置信区间统计结果
在采用 2×12 的模拟预试验的方式进行模拟计算的结果显示同一个参比制剂其 CVW 值分别在 22.7% ~ 49.7%(R1,Cmax)、17.7% ~ 64.8%(R2,Cmax) 和 17.6% ~ 51.4%(R1,AUC)、19.1% ~ 58.3%(R2,AUC)这样一个比较宽泛的内波动,统计这些数字在不同区间内出现的频次见图 1。从图可见,CVW 值在 26% ~ 50% 之间的分布相对而言在各个区间没有太明显的区别,同一个参比制剂分别按照 R1 和 R2 来进行统计的的结果还是有一定的差异的,这充分的说明了预试验所取得的CVW 值除了在个别的区间内出现的几率略高一些外,在很宽的一个范围内出现的几率相差并不大。并且,模拟预试验的计算结果中出现了相当一部分(最少的有 14 次)的 CVW 值是小于 30%的这样的结果,这进一步提示了我们预试验的结果很难用于对正式试验的CVW 值的预测,因此,用这样的结果来预测正式试验的例数是不可靠的。
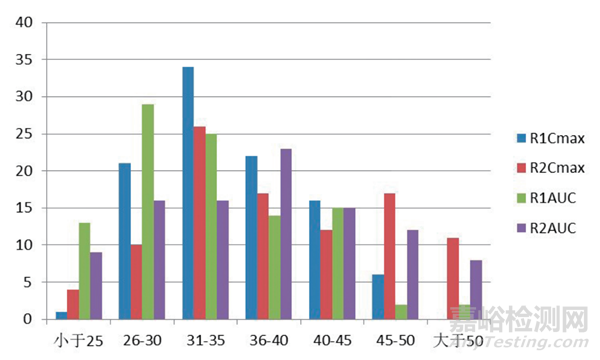
图1 参比制剂主要药代动力学参数个体内变异系数概率分布图
在统计 100 次模拟预试验所取得的 Cmax 和 AUC 的几何均值的结果见图 2、图 3、图 4。从图可见,对于同一参比制剂的 2 次给药的模拟计算结果而言,无论是 Cmax 还是 AUC 的比值,都分布在一个比较宽的范围之内,因而,Cmax 和 AUC 即使对于同一个药物而言,它的几何均值比在一个比较宽的范围内普遍分布,单次的预试验取得的 GMR 的数值的随机性是比较大的,用来计算正式试验需要的试验例数自然也会有很大的随机性。值得注意的是,这些均值比绝大多数情形下都分布在 0.80 ~ 1.25 之间,这从某个层面也可能证实了采用生物等效标准的范围为80% ~ 125% 是合理的。
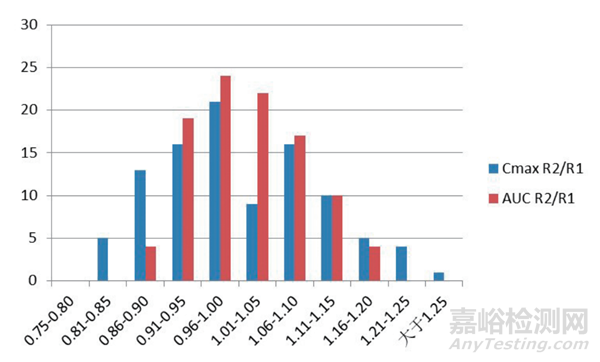
图2 参比制剂药代动力学参数(Cmax、AUC)几何平均值概率分布图
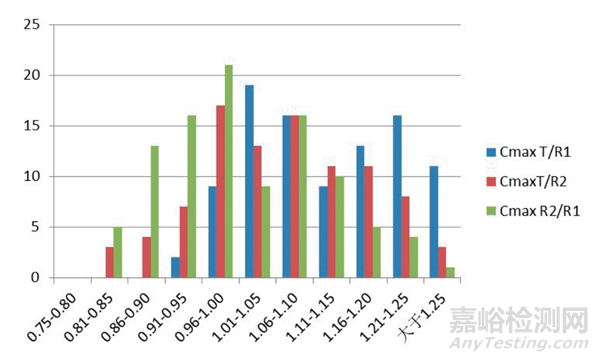
图3 参比制剂/受试制剂药代动力学参数(Cmax)几何平均值概率分布图
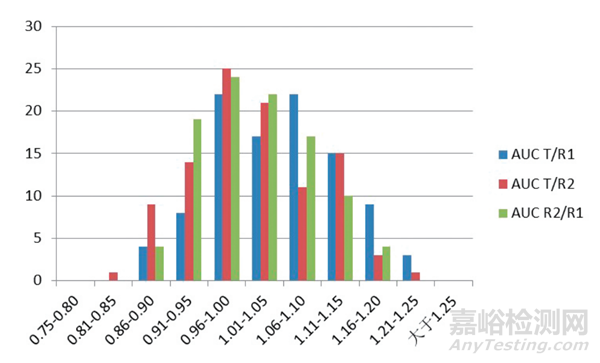
图4 参比制剂/受试制剂药代动力学参数(AUC)几何平均值概率分布图
从统计两次参比制剂和受试制剂的药代数据的几何均值的结果中可见,无论是用哪一次参比制剂数据来进行处理,几何均值比都是随机出现在 0.86 ~ 1.25 之间非常宽的一个范围之内,并且会有相当一部分结果超出了1.25 的范围。因此,单个预试验的几何均值比的结果实际上还是在比较大范围内的一个随机数,因此用这个数值来计算受试例数,会有很大的风险。
2.2 用于制剂处方工艺开发的风险分析
有很多专家学者认为可以从 BE 预试验的结果中来定性判断受试制剂和参比制剂相比的暴露量的相对大小,即预试验的几何均值如果大多数情形下 T/R大于 1,或者小于 1 即说明数据有单边偏向,认为受试制剂的暴露量比参比制剂大或者小,并以此为依据进一步进行制剂的处方和工艺的优化工作。
但是我们从以上的几个图表中可以明显地看到这些几何均值的分布在一定范围内的分布是随机的。因此,预试验并不能真正体现受试制剂和参比制剂药物暴露量的相对大小。为了更加准确地说明这个问题,排除由于单个受试者过高或过低的血药浓度的试验数据带来误差的影响,我们分别统计了每一例受试者的受试制剂和参比制剂的 Cmax和 AUC 的几何均值的比值的大小,对于在正式的试验中 Cmax 的几何均值比 T/R1、T/R2 分别有 24 个受试者和19 个受试者是大于 1,而 AUC 的几何均值比 T/R1、T/R2 中则分别有 21 例和 15 例受试者大于 1,说明除了 T/R2的 AUC 的比值相对而言有一定的偏向,而其他的数据没有单边偏向。我们进一步统计每一次预试验的结果,统计每一次受试制剂和参比制剂的比值大于 1 或者小于 1 的情形,结果见图 5 和图 6,从图可见每一次模拟预试验抽取的样本中,出现受试者使用参比制剂和受试制剂都会出现比较多的“单边”的现象,100 次中,12 例出现仅出现 3 例受试制剂暴露量超过参比制剂的几率和出现 9例受试制剂暴露量都超过参比制剂的几率相差不大。因此,虽然从总体上来看有一定的集中度,但是单次预试验的结果的随机性还是比较大,从预试验中取得的 T/R>1 的数据来分析受试制剂和参比制剂的暴露量的关系也是有很大的风险的,用这样的结果来指导制剂处方工艺开发存在较大风险。

图5 参比制剂/受试制剂药代动力学参数(AUC)几何均值概率分布图
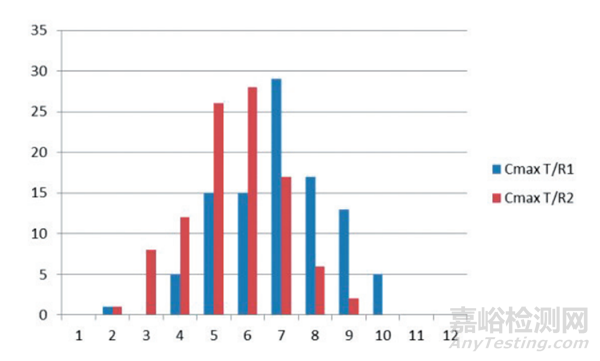
图6 参比制剂/受试制剂药代动力学参数(Cmax)几何均值概率分布图
在实际操作过程中,我们不可能去做多次的预试验来预测正式试验的结果以及相关的试验设计,通过模拟计算发现单次的试验的 Cmax、AUC 的几何均值和 CV 值的在一个比较大的范围内是随机出现的。而且,通过模拟计算也表明所谓的数据“单向偏移”存在着同样的概率出现,预试验的结果也不能给出一个定性的受试制剂和参比制剂暴露量之间的关系的方向。
3、讨 论
由于高变异药物受个体内变异影响较大,通过两周期交叉试验设计法生物等效性预试验对正式试验样本量估计以及指导制剂处方工艺开发,存在较大风险,因此不建议采用两周期交叉试验设计对高变异药物进行生物等效性预试验研究。
由于仿制药与参比制剂处方工艺存在一定差异,文献报道数据仅供参考,在制剂处方工艺开发阶段及正式生物等效试验开展前,药物研发机构最好开展预试验研究,以降低研究成本及正式生物等效性的风险。在如何高效地进行高变异药物的生物等效预试验的研究过程中,可采用重复试验设计法(部分重复或完全重复)或两阶段设计法。
国家药品监督管理局发布的《高变异药物生物等效性研究技术指导原则》[1],为以药动学参数为主要终点指标的高变异化学药物开展生物等效性试验研究设计、样本量估算、统计分析、结果报告等方面提供了技术指导,建议采用重复试验设计(部分重复或完全重复)进行生物等效试验预试验,获得必要的药代动力学参数,为正式生物等效试验提供数据支持,设计合理的临床研究方案,并确定合理的样本量。
近年来,两阶段设计(Two-StageDesign,TSD)生物等效性研究由于其在样本量估算方面的适应性、灵活性,已被各国指南所认可(FDA、EMA、加拿大、澳大利亚、日本、新西兰、WHO 等)[6]。虽然在《中国药典》9011 药物制剂人体生物利用度和生物等效性试验指导原则中明确指出:“在证明生物等效性时,可以接受两阶段试验方法,最初一组受试者给药并分析数据,如果不能证明生物等效,则可以增加招募一组受试者,在最终分析中合并两组的结果”,制药企业及药物研究单位可在生物等效预试验研究中参考使用。
参考文献
[1] 美国食品药品监督管理局,2013,以药代动力学为终点评价指标的简化新药申请的生物等效性研究的指导原则.
[2] 于永沛,闫小妍,姚晨等. 高变异药生物等效性研究中的样本量估计 [J]. 中国新 药杂志,2018,27(9):1019-1024.
[3] 国家药品监督管理局,2018,高变异药物生物等效性研究技术指导原则.
[4] Isabel Moreno1,Dolores Ochoa,Manuel Roman, et al. Utility of PilotStudies for Predicting Ratios andIntra-subject Variability in HighVariability Drug[J].Basic & ClinicalPharmacology & Toxicology,2016.
[5] 刘甜甜,陆梦洁,周潼潼等,高变异药物生物等效性评价确切样本量计算 [J]. 中国临床药理学杂志,2017, 33(6):1152-1156.
[6] 孙华,谢大虎,梁海棠,两阶段生物等效性研究的试验设计及关注要点 [J]. 药物评价研究,2017,40(5):593-599.
本文作者刘清梁1,3、王文峰2、邹江1、杨琰1、张吉喜3*,1华润双鹤药业股份有限公司、2华润医药集团、3重庆大学生物工程学院,仅供交流学习。

来源:制药工艺与装备


