您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-11-30 19:27
摘 要 / Abstract
随着医疗器械领域的快速发展,传统随机对照试验在评估医疗器械有效和安全性时面临诸多挑战。全球医疗器械监管机构开始探索并采纳真实世界证据(RWE)作为医疗器械监管决策的重要依据。本文对2012~2019 年美国食品药品监督管理局(FDA)利用RWE 进行医疗器械临床评价的90 个案例进行系统分析,重点聚焦于不同审批途径、风险等级产品、疾病领域以及注册阶段RWE 的特性。结果表明,近半数产品(44.4%,50/90)将RWE 作为注册审批过程中的主要或唯一证据。这些产品主要注册目的集中在上市前批准,疾病领域为心血管疾病,医疗器械类别为Ⅱ类和Ⅲ类。RWE 在医疗器械监管决策中越来越重要,尤其是在中低风险医疗器械的上市前审批中。
With the rapid development of medical devices, traditional randomized controlled trials (RCTs) face many challenges in evaluating their effectiveness and safety. Regulatory agencies worldwide have begun to explore and use real-world evidence (RWE) as an important basis for medical device regulatory decision-making. This study conducted a systematic analysis of 90 cases of clinical evaluation of medical devices using RWE by the US FDA between 2012 and 2019. It focused on the characteristics of RWE across regulatory pathways, risk classes, disease areas, and stages of approval. The study found that nearly half of the products (44.4%, 50/90) used RWE as the primary or sole evidence in the premarket approval process. These products were predominantly focused on premarket approval, within the cardiovascular disease domain, and consisted mainly of Class II and Class III devices. RWE is becoming increasingly important in medical device regulatory decision-making, especially in the premarket approval of low-to-medium risk medical devices.
关 键 词 / Key words
真实世界数据;真实世界证据;医疗器械;注册上市
real world data; real world evidence; medical devices; registration and approval for marketing
严格的临床证据是医疗器械上市注册的重中之重, 传统随机对照试验(randomized controlled trial,RCT) 一直被视为评估医疗干预的金标准[1]。但随着医疗器械产业的高速发展,传统的RCT 常因医疗器械的复杂性、多样性、伦理等因素难以实施[2]。鉴于传统RCT 在医疗器械评估中的局限性,各国和地区医疗器械相关监管部门与研究人员积极探索新的证据来源支持医疗器械临床评价。随着大数据时代的到来,许多国家和地区的监管机构越来越重视真实世界数据(real world data,RWD) 与其产生的真实世界证据(real world evidence,RWE) 在医疗器械全生命周期监管中的作用[3]。
美国食品药品监督管理局(Food and Drug Administration,FDA) 率先在国际上探索利用RWD 和RWE 开展药械临床评价,并发布了一系列指导原则[4-5]。2021 年,FDA 发布了90 个2012~2019 年通过RWE 开展医疗器械监管决策的成功案例[6]。本文通过对这些案例进行总结,以发现RWE 作为主要或唯一证据支持医疗器械上市注册的特点, 为我国RWE 支持医疗器械监管决策提供参考与借鉴。
1、方 法
1.1 数据来源
本文数据来源于2021 年FDA 发布的90 个自2012~2019年通过RWE 开展医疗器械监管决策的成功案例。
1.2 统计分析方法
本研究由流行病学、统计学、临床医学等多学科专家组成的团队,结合既往相关监管部门发布的指导原则,建立了标准化的数据提取表。数据提取表主要包括:产品基本信息、数据库构建、研究设计与统计分析、偏倚控制等。采用文献计量分析法,对90 个案例中的公开文件进行检索。相关信息由经统一培训的研究人员, 以背靠背的方式, 再结合Pubmed、Embase 等数据库以及 clinical trials.gov 等临床试验注册网站检索产品的注册信息和研究成果,按照数据提取表提取数据。
本文主要为描述性分析,对于连续性数据,采用均值标准差或者中位数四分位数(Median,IQR) 进行描述;对于分类数据,采用频数及构成比的形式进行描述。采用数据提取软件为Epidata 3.1,统计分析软件为R4.22。
2、结 果
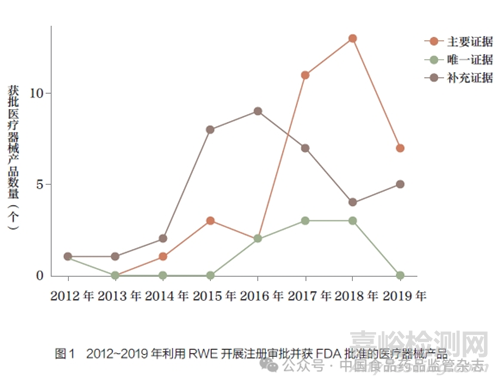
在90 个案例的所有产品中,通过上市前批准(premarket approval application,PMA)路径注册审批的为61.1%, 且均为Ⅱ 和Ⅲ 类医疗器械产品,62.2% 的产品有已上市的同类产品,56.7% 的产品为心血管疾病类医疗器械。51.1% 的产品将RWE 用于上市前的注册审批,20.0% 的产品在全生命周期利用RWE 开展审批。2016 年后获得FDA 批准的数量较多,2017 年和2018 年获批产品的比例分别为23.3% 和22.2%,如图1 所示。在RWD 类型的选择上,61.1% 和27.8% 的产品所采用为登记数据库和电子病历数据库。37.8% 的产品样本量在100~1000 人。产品注册以上市前批准(17.8%) 和适应症/ 说明书修改(16.7%)为主,见表1。
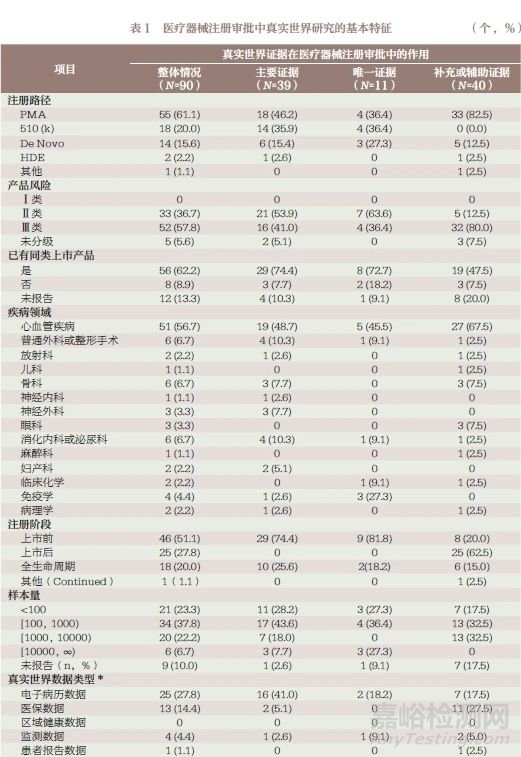
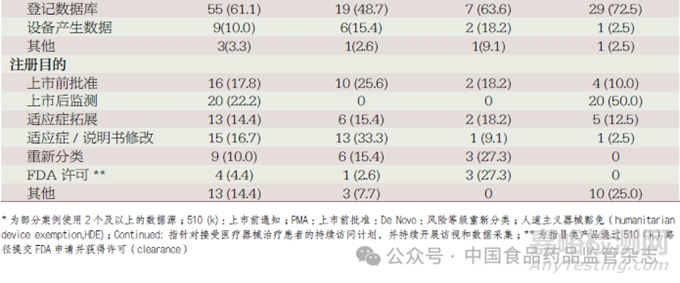
在FDA 发布的90 个真实世界研究案例中,有50 个产品将RWE 作为注册审批过程中的主要或唯一证据。由表1 可知,将RWE 作为主要证据提交审批的39 个医疗器械产品中,14 个(35.9%)产品通过上市前通知[510(k)] 路径进行注册审批,18个产品(46.2%) 通过PMA 进行注册审批。Ⅱ类医疗器械产品数量有21 个(53.8%),Ⅲ类医疗器械产品数量16 个(41.0%)。74.4% 的产品均有已上市的同类产品,48.7% 的产品适用于患有心血管疾病,普通外科或整形手术和消化内科或泌尿外科的产品比例均为10.3%。74.4% 的产品RWD 仅用于上市前的递交。2017 年,FDA 发布关于应用RWE 进行医疗器械审批的指导原则,批准的医疗器械数量增长迅速,71.8% 的产品样本量小于1000 人。48.7% 和41.0% 产品的数据来源类型分别为登记数据库与电子病历数据库。33.3% 和25.6% 的产品分别用于适应症/说明书修改和上市前批准。
由表1 可知,将RWE 作为唯一证据提交审批的11 个医疗器械产品中,4 个(36.4%)产品通过510(k) 进行注册审批,4个(36.4%) 通过PMA 进行注册审批。Ⅱ类产品和Ⅲ类产品数据分别有7 个(63.6%)、4 个(36.4%)。72.7% 的产品均有已上市的同类产品,45.5% 的产品为心血管疾病医疗器械。81.8%的产品仅用于上市前的递交,88.9% 的产品2017 年后才被FDA 批准。63.7% 的产品样本量小于1000 人,27.3% 的产品样本量大于10000 人。采用登记数据库、电子病历数据库及设备产生数据的比例分别为63.6%、18.2%、18.2%、27.3% 的产品用于风险等级重新分类(DeNovo),510(k) 注册的产品比例为27.3%。
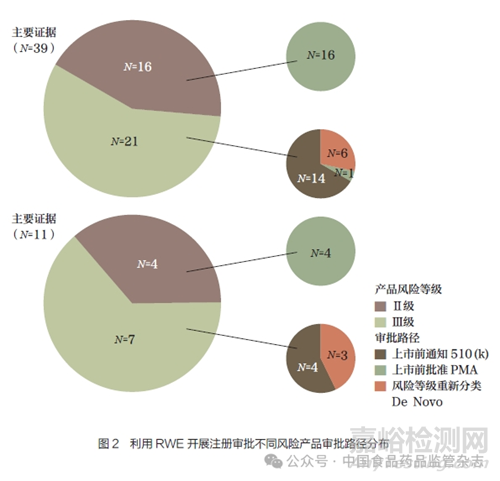
在RWE 被作为主要或唯一证据的产品中,100% 的Ⅲ类医疗器械通过PMA 的方式进行注册审批。而Ⅱ类医疗器械产品则主要通过510(k)和De Novo 的路径进行注册审批,如图2 所示。
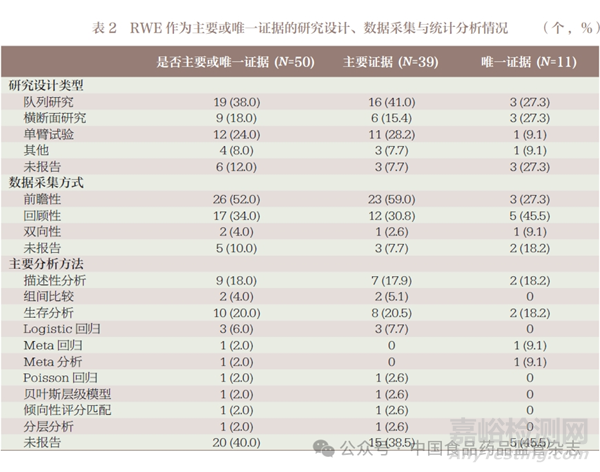
使用RWE 作为上市前主要或唯一证据来源的50 例产品中,队列研究19 例(38.0%)、横断面研究9 例(18.0%)和单臂试验12 例(24.0%),6 例(12.0%)研究未报告研究设计类型,其他的研究设计类型还包括成组序贯设计、Meta 分析、多臂研究等。在统计分析方面,20 个产品没有报告使用的统计方法,其余的研究中有9 个(18.0%)案例采用描述性分析的方法,计算了各组的人数和构成比,但没有进行组间比较和假设检验,2 个产品采用Fisher 确切概率法和Wilcoxon 秩和检验进行组间比较。10 个产品采用生存分析的方法,包括Cox 回归、竞争风险分析、 Kaplan Meier 方法等,还有1 项研究结合使用倾向性评分匹配的方法。使用Logistic 回归分析的有3 个产品,其中1 个产品研究结合使用广义估计方程。使用Meta 回归、Meta 分析、泊松回归、贝叶斯层级模型、倾向性评分匹配和分层分析的方法各有1个产品,见表2。
3、讨 论
从以上分析结果可以看出,FDA 在开展医疗器械审批注册时,RWE 作为证据来源的重要性越来越凸显。2021 年FDA 发布的90 个案例产品中,近一半的审批产品都以RWE 作为主要或唯一证据,且广泛利用RWE 开展上市前批准、适应症拓展、适应症/ 说明书修改等注册目的。在这些产品中,56.7% 的医疗产品针对心血管疾病,而在RWE 作为主要或唯一证据的分类中,有24 例(48.0%)的产品用于心血管疾病,这些医疗器械多集中于以下几类:冠状动脉支架系统(4例,16.7%)、经皮血管内成形术球囊导管(6 例,25.0%)、心脏瓣膜(3 例,12.5%)、除颤器(5例,20.8%)等,均属于临床实践过程中应用较多的医疗器械种类。全球疾病负担数据(Global Burden of Disease 2021)显示,心血管疾病在美国的死亡率排至第1 位[272.26/100 000,95%CI:(236.18/100 000 , 290.68/100 000)],针对心血管疾病的医疗器械研究较多,发展相对成熟,数据资源更加丰富,有助于进一步开展真实世界研究。
90 个案例产品中主要为Ⅱ类和Ⅲ类医疗器械,提示FDA 已开始利用RWE 进行此类医疗器械的审批探索实践。但在Ⅲ类医疗器械中,RWE 更多地被作为补充证据用于上市后监测和适应症拓展,而且这些Ⅲ类医疗器械均通过PMA 的路径递交,需要利用RWE 提供新的科学证据。在Ⅱ类医疗器械中,较多的通过510(k)和De Novo 的路径递交。由此可见,目前FDA 对Ⅲ类医疗器械利用RWE 开展注册审评较为谨慎。
支持监管决定的RWE 强度取决于临床研究方法和基础数据的可靠性和相关性,而数据的可靠性则需要对数据获取和数据质量控制进行评价。在质量得到保证时,从这些数据生成的证据可提供有关医疗器械有效性和安全性的相关信息。从分析结果来看,被作为主要或唯一证据的RWE中,多为前瞻性收集的数据,电子病历数据和登记数据被应用的最为广泛,登记数据的比例略高于电子病历数据。2017 年FDA发布的指导原则中,认为特定的注册数据库可能更适合用于上市后监测,不足以支持合理保证安全性和有效性或实质等同性的上市前确定[4]。但在本次发布的90个案例产品中,有由申办方或第三方机构建立的登记数据库在上市前的注册审批中作为主要或唯一临床证据来源的案例,其中一个产品通过链接国家登记数据库和保险数据库,获得了长达5 年随访期的结局数据,成功获得了FDA 的上市前适应症拓展审批。
由于RWD 存在人群异质性、数据的多样化、数据标准不够统一的特点,因此数据采集及其质量控制非常重要。从上述分析结果可以看出,将RWE 作为主要或唯一证据时,多采用前瞻性的数据采集方式;在研究设计方面,这些案例以队列研究、单臂研究和横断面研究较为常见。在统计分析方面,通过公开文件未能获取大部分研究的细节,但从可获得的信息中可以发现,对相关结局进行描述的RWE 占到了相当的比例。这些以描述为主要分析方法的研究,涉及的医疗器械产品种类较多,与心血管疾病相关的医疗器械(4 个),多为手术操作过程中的辅助器械,例如液氮冷冻治疗手术系统、辅助控制腹腔镜仪器系统等。
2020 年3 月26 日, 青光眼引流管注册申请成功通过审查,成为我国首个使用境内真实世界数据获批的医疗器械产品[7],为我国后续利用RWE 开展医疗器械审批起到了先行先试的作用[8]。目前,海南博鳌乐城国际医疗旅游先行区(以下简称乐城先行区)共11 款医疗器械产品获得国家药监局批准注册上市,其中RWE均作为辅助证据,即在已有的境外临床试验结果的基础上,用于补充我国人群有效性和安全性证据;或在同品种路径中通过真实世界研究(real world study,RWS)论证申请产品与同种产品的差异部分的安全性和有效性;或通过开展国内临床试验加乐城真实世界证据。在乐城先行区开展的RWS 均为境外已上市而国内未注册的创新医疗器械,以上产品通过“乐城模式”开展临床评价的路径主要包括以下3 种:①目标产品境外临床试验数据+乐城RWE ;②国内同品种已上市产品+ 目标产品乐城RWE ;③ 目标产品国内临床试验+ 乐城RWE。目前,在乐城开展的RWS,在研究前期均会构建多学科的专业团队,在完善的研究设计框架和高质量的数据采集和管理条件下,通过构建高质量的登记数据库,支持产生高质量的RWE[8-10]。
4、结 语
目前,我国医疗器械的监管正处于创新转型的阶段, 其中RWD 与RWE 为监管提供了更广泛的证据来源,本文通过总结FDA 发布案例情况,结合现有的政策背景,以期能推动监管部门持续RWD 与RWE 的创新应用,建立更加完善、科学的体系,通过更加灵活的评审策略,推动我国医疗器械产业的发展。
引用本文
王鸣岐,介万,任燕,胡莎,云虹伟,陈淑文,林凯*,姚明宏*,孙鑫*.FDA 采用真实世界证据作为主要或唯一证据支持医疗器械上市注册的产品特征分析[J].中国食品药品监管,2024(10):68-75.

来源:中国食品药品监管杂志


