您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-02-14 08:51
将新疗法推向市场的迫切愿望,促使大型制药公司、生物技术公司和CRO部署AI/ML技术,以加强和加快药物研发进程。这些公司面临着“自建”还是“购买”的抉择,要么投资内部员工和基础设施,建立内部能力,要么与人工智能公司合作。
2025年1月8日,来自CRO公司Evotec的多位专家在Journal of Medicinal Chemistry上发表文章Real-World Applications and Experiences of AI/ML Deployment forDrug Discovery,以CRO的身份阐述了他们的观点。他们认为,将成熟的计算方法、人工智能/ML 技术和人类经验相结合,可以产生最佳结果。
概要
深度学习的方法现在正在影响药物发现过程的每个阶段,从早期靶点识别到苗头化合物发现和先导化合物优化。
然而,由于生物系统固有的复杂性、高质量训练数据的可用性以及化学描述符全面捕捉化学相互作用本质的能力有限,准确预测实验数据仍然具有挑战性。此外,药物发现决策中固有的偏差已被充分记录。这些偏见会阻碍进展,妨碍人工智能/ML技术的整合。关于AI/ML技术的有效性及其对加速药物发现过程的影响的说法常常被夸大,这使得情况变得更加复杂。
根据Evotec的经验,将成熟的计算方法、人工智能/ML技术和人类经验相结合,可以产生最佳结果。这里简要总结了Evotec的专家和其他人在AI/ML应用方面的经验,这些应用目前对Evotec的工作影响最大。
AI/ML技术在药物发现中的应用
化学空间的机器表征
使用深度学习来表征化学空间是化学信息学的最新重大发展。化合物现在可以由向量表征,向量是通过在大型化合物数据库上训练深度神经网络生成的。这种表征被称为潜在空间,因为它们是从数据集中以数学方式推导出来的,并封装了其基本特征。给定的向量(在这个潜在空间中的位置)可以解码为化学结构,这比分子指纹等旧表征形式有很大的好处。它能够快速鉴定新区域中的目标化合物。例如,向量之间的插值允许探索中间化学结构,这可能是进入可申请专利的化学空间的一种方式。
一个开创性的例子是连续和数据驱动描述符(CDDD),我们已广泛用于生成化合物设计(请参阅下面GenerativeDesign部分中的其他方法)。CDDD是一种自动编码器(AE),同时在SMILES上进行训练并受化学性质(例如极性表面积和亲脂性)的约束,这些化学性质将化学和物理相似的分子推入相似的潜在子空间。这种训练方式倾向于迁移学习(TL)的表征,即通过添加新的、特定于项目的训练数据来改变预训练模型的任务,从而专注于特定于项目的目标和化学性质。这种AE架构提供的分子相似性和计算特性的联系是相对于指纹图谱的另一个优势。
我们开发了自己的基于AE的内部Seq2Seq模型,利用递归神经网络(RNN)和transformer架构。通过在内部策划的数据集上训练这些模型,我们提高了下游任务的性能和灵活性。改进包括覆盖分子量大于600 Da的化合物,这对于某些项目来说是必需的。它们还包括提取分子的潜在特征,用于定量构效关系(QSAR)模型构建。将QSAR和深度生成化学(DGC)结合在同一潜在空间中,我们采用贝叶斯优化(BO)等优化算法和粒子群优化(PSO)执行反向QSAR/inverse设计。这意味着我们可以生成针对QSAR模型预测进行优化的化合物设计。
这些表征的质量至关重要,因为它直接影响后续应用程序的可靠性和准确性。我们根据DGCSMILES的有效性、新颖性和药物相似性,以及量化QSAR性能和潜在空间目标函数平滑度的指标来验证我们的表征模型。总之,这些验证使我们的科学家能够做出明智的决策并自信地构建ML模型。
机器学习
在本节中,我们将简要介绍如何使用ML来预测活性和吸收、分布、代谢、排泄和毒性(ADMET)终点和化合物的物理化学性质直接来自分子结构─方法通常分别称为QSAR和定量结构-性质关系(QSPR)建模。
预测模型的质量取决于训练数据的质量。我们的实验数据通过标准化分析生成,经过精心策划,以去除不可靠或不一致的测量结果。这些检测包括logD、水溶性、Caco2通透性、微粒体清除率和hERG通道抑制。为回归(连续预测)和分类任务(离散预测)实施了特定的管理流程,确保仅使用高质量的数据。为了简化ML活动并促进模型的定期训练和更新,我们实施了自动化ML工作流程,其中包括化学结构准备、描述符计算、模型选择、超参数优化和模型交付。ML生成的预测最终使用可解释性技术进行解释,该技术估计输入特征对模型决策的贡献。
近年来,深度学习技术在QSAR/QSPR建模中的应用显示出巨大的前景。特别是图形神经网络(GNN),已被证明在某些端点上优于随机森林(RF)等传统ML算法。然而,根据我们对数据集通常跨越几百到一万多个数据点的经验,传统的ML算法通常优于深度学习模型。尽管如此,GNN已被证明在应用于更大的数据集时有助于提高模型的性能和稳健性。
预测性QSAR和QSPR模型在发现项目中发挥着关键作用,有助于化合物想法的选择和优先级排序。在这种情况下,一个应用程序是我们的生成工具的评分函数。
生成式设计
使用DGC设计具有靶向特性的化合物最近已成为药物化学中的一种强大方法。我们之前的评论确定了2017年至2020年间发布的100多种深度学习从头设计方法。从那时起,人们对这个话题的兴趣激增,使得跟踪所有新文章变得困难。我们发现这些论文通常缺乏实际应用的视角,因为许多研究人员没有幸运地能够综合和测试他们的设计。我们利用我们的机会,定期成功地使用最先进的2D和3D DGC工具来设计化合物,然后进行制造和测试。
我们根据内部反馈采用和修改的一个工具是REINVENT。这是一种强化学习方法,它使用正反馈循环生成得分更高的化合物设计。我们的研究结果表明,它产生相关分子的能力可以与用于推动项目特定目标的评分组件高度相关。特别是,与单独使用2D分数相比,基于药效团的匹配或对接分数等3D组件在所需化学空间中生成设计的速度要快。在随后的迭代中,可以使用物理化学性质和ADMET端点的高级QSAR模型以及更标准的计算化学工具来改善生成的化合物的性质。
对任何生成工具获得的结果进行后处理都至关重要,主要有三个原因。首先,由于某些评分组件的固有计算成本,它们只能后验使用。这些评分方法的示例包括相对结合自由能(RBFE)和片段分子轨道(FMO)相互作用能。其次,深度生成工具不能总是同时优化多个组件,因此,其中一些组件必须在后处理阶段按顺序应用。例如,在口袋内培养配体的方法通常侧重于焓对效力的贡献,例如蛋白质-配体相互作用。最后,药物化学项目会随着时间的推移而发展,目标化合物的性质也会随之发展。鉴于此后处理步骤的重要性,我们正在开发自动化管道,以集成传统的计算化学、AI/ML和基于物理的计算,以加快这一过程(请参阅下面的计算管道)。
蛋白质建模
准确的蛋白质模型对于药物发现项目非常有用。通常,此类模型是使用X射线晶体学或低温电子显微镜(cryo-EM)等实验方法获得的。直到最近,只有非AI方法被用于构建蛋白质的同源模型,而这些模型没有实验模型。然而,最近,利用人工智能预测蛋白质结构的方法家族的一员AlphaFold2(AF2)在其预测中表现出了非凡的准确性。我们的本地安装是生成用于迭代蛋白质构建设计和准备模型以拟合实验获得的密度的重要资源。我们结合了AF2和ProteinMPNN提高蛋白质稳定性和产量。这种方法可以改变只能分离极少量蛋白质的项目。AFMultimer的能力预测蛋白质-蛋白质复合物的3D结构有助于结构生物学家获得靶点的初始模型。此类模型可以拟合到实验密度中并进一步细化。可以使用FoldDock对新型复合物进行建模,它优化了AlphaFold多聚体运行的多个序列比对,根据区分可接受模型和错误模型的分数产生更好的预测。
AlphaFold DB数据库由DeepMind提供并由EBI托管的AF2模型数据库,结合我们安装的AFMultimer,是药物设计许多方面的巨大资源,从靶点配体估计到VS和对接。然而,我们的目标是在与目标配体的复合物中为我们的药物靶点构建我们自己的实验结构。当这是不可能的时,我们通常在已知配体存在的情况下使用经典方法构建同源模型,以便结合位点中的侧链处于适合对接的构象。
深度学习的最新进展也使配体-蛋白质复合物的预测方法成为可能。RoseTTAFold-AllAtom,Umol和AF3等方法声称可以预测靶蛋白与小分子配体、金属离子、核酸和共价结合剂相互作用的结构细节,其精度超过了已建立的对接方法。我们以极大的兴趣关注这一领域的发展。
主动学习
药物化学通常在有限的实验数据下运作。对于致力于新靶点的项目的苗头化合物到先导阶段尤其如此。在数据稀少且生成成本高昂的情况下,主动学习(AL)可能非常有用,因为它的目的是以最有效的方式生成足够的数据。准确地说,AL是一种基于ML的策略,旨在以最少的数据最大限度地提高特定任务(目标函数)的学习性能。该算法根据所谓的获取函数从预定义的未标记项目池中迭代选择,该函数平衡了开发(根据当前知识选择最有前途的项目)和探索(从化学空间中不太已知或未知的区域中进行选择,以增强模型的整体知识)。
类似地,BO试图在完全定义的参数空间内确定下一个要测试的化合物,以找到目标的最佳在这种情况下,这可能是多参数优化(MPO)分数。这些MPO评分可以包含具有更多数据点(如效力、亲脂性、代谢稳定性和通透性测量)的主要检测成分,也可以包含具有较少数据点(如针对酶、受体和转运蛋白的脱靶活性)的后续检测,具体取决于项目要求。在药物化学中,AL用于指导从广阔的化学空间中选择信息丰富的化合物。我们使用AL来实现超大型按需化合物库(如EnamineREAL)的VS并减少实现项目目标所需的化合物数量。
传统的基于结构和基于配体的方法对于数十亿种化合物的暴力筛选来说,计算成本太高且耗时。此外,VS成本随着评分函数的复杂性而增加。我们的解决方案基于开源MolPal构建,将BO与VS工具和基于高级分子动力学(MD)的评分函数相结合,将探索重点放在性能最高的化合物上。
设计-制造-测试-分析(DMTA)循环可以配置为探索化学空间的AL过程。我们以这种方式使用BO,通过选择要制造和实验验证的化合物来协助决策过程。像这样信息丰富的化合物的选择最终应该会导致循环次数的减少。在其AL形式中,BO对来自其他工具或药物化学家想法的预定义化合物列表进行排名。虽然这种方法限制了探索能力,但它可以提高药物设计师对所提出解决方案的接受度,并将搜索空间减少到更易于管理的大小。在其生成形式中,BO提出了新的点,以在基于机器的化学空间表征中进行测试(见上一节)。建议的点必须解码为化学结构。这些设计可以挑战团队的心态并避免不必要的人为偏见。然而,它们并不总是很容易合成。药物化学团队的反馈可以突出改进的协同机会,例如,标记来自单个异常结果的多个设计以及来自合成改进的新机会。
合成可追溯性和逆合成预测
化合物的合成或“Make”阶段通常是DMTA循环中的限速步骤。因此,合成可处理性是“设计”阶段的一个关键方面。这适用于人类和AI生成的设计。目前,大多数生成式设计工具并未在其用于生长或评分化合物的算法中明确编码此标准。然而,该领域最令人兴奋的发展之一是AI计算机辅助综合规划(CASP)工具的发明。这使得使用成熟的逆合成分析或更快的ML模型通过合成可处理性进行评分或过滤成为可能。药物化学家通常在设计化合物时考虑合成路线,或者至少在脑海中估计所涉及的难度。
最先进的AI工具尚未达到药物化学家团队每天分享专业知识和知识的复杂程度和效率,例如关于构建砌块和中间体的可用性和反应性。但是,添加内部数据,例如来自电子实验室笔记本(ELN)和积木库存的数据,确实提高了工具的有效性。AI逆合成越来越多地被药物和计算化学家使用,例如用于骨架跳跃、灵感和更轻松地规划简单路线。与其他领域的AI一样,如果期望与用户自己的专业知识和特定经验相当,逆合成输出可能会给人留下令人失望的第一印象。我们的化学家通过Web界面使用商业AICASP工具来获得灵感或交叉检查他们的路线规划;他们发现其指向背景文献的快速简便的链接非常有用。事实证明,评估工具(其中一些非常昂贵)对我们来说很困难,这可能是因为我们对性能的期望不切实际。对于生成式设计工作流程,ML合成复杂性分数具有一些实用性,但我们始终将合成可追踪性的手动评估作为最后步骤之一。
安全性评估
除了合成的可处理性外,还必须考虑给定化合物设计的安全风险。安全性仍然是药物开发项目的主要关注点。通常,只有在部署了大量资源后,安全风险才会在药物开发的后期变得明显。因此,越来越多的AI/ML方法可以更早、更便宜地发现安全风险,受到相当大的关注。例如,人们已经开发了纯计算机模型,以降低药物诱导的肝损伤(DILI)的可能性,基于化合物描述符(如sp中的碳原子数)杂交。计算机模拟模型是可取的,因为它们可以在合成化合物之前帮助设计,从而可能降低与探索性安全性分析相关的成本。这些模型往往是基于规则的,或者采用传统的监督式ML算法。然而,为了提高预测性能,结合体外数据(例如,胆盐输出泵(BSEP)转运蛋白抑制和细胞毒性数据)来构建更复杂的系统是有益的,例如贝叶斯模型。
与仅涵盖有限毒性方面的个体体外检测相比,组学技术提供了响应药物暴露的细胞状态的更全面快照。幸运的是,新的高通量组学技术允许创建足够大小的数据集来训练AI模型。这些模型可以识别组学谱中与导致器官毒性的不良结果相关的复杂模式。经过训练后,他们可以高精度地预测新化合物的毒性风险,优于现有的体外方法。此外,这种方法不仅限于小分子,而且同样适用于包括生物制剂在内的其他形式。为了为我们的AI模型创建训练数据集,我们利用我们的高通量转录组学平台(ScreenSeq)生成了一个从细胞模型获得的转录组学图谱数据库。由数百种表征良好的不同类型的化合物生成的曲线可作为有用的参考点。
计算管道
从头设计方法的出现,尤其是深度生成式AI方法,增加了对大量虚拟化合物进行评估和优先排序的需求。这通常是通过将预测模型与更简单的计算属性和/或更复杂的基于物理的分数一起应用来实现的。根据多个标准(药物相似性、预测活性和ADMET属性、新颖性、物理化学性质、合成可追踪性等)对每个虚拟分子进行评分,然后使用临时的、项目特定的MPO函数对不同的分数进行汇总。正确参数化后,该MPO分数可用于对虚拟分子进行排名,并为下一轮合成优先考虑最有前途的化合物。部署此类管道时的一个技术挑战是不同任务之间的编排,因为通常涉及的工具数量和多样性。一个好的编排器需要能够在不同的文件格式之间进行交互,处理多个环境,有效地管理资源,在需要时扩展作业。由于AI/ML领域正在迅速发展,因此还需要设计出能够轻松添加新组件或更改部署它的基础设施的方式。
DMTA循环的自动化可以节省时间和资源,同时编码最佳实践并提高可重复性,这有助于在选择合成设计时保持客观性。有好几个商业和开源平台在设计时考虑了自动化药物设计。我们深受Green和Besnard等人工作的影响。并寻求尽可能使用Knime或我们内部的高性能计算(HPC)流水线解决方案来自动化我们的工作流程。我们面临着与BRADSHAW作者相同的挑战集成性、稳健性、简单性和灵活性。每个管道都需要适应项目不断变化的需求,同时至少部分可被其他项目重用。
药物化学项目背景下的AI
AI设计工具的出现,加上基于物理的方法的日益影响和HPC成本的降低,促使一些制药公司探索不同的工作方式。在Evotec,我们有一个AI/ML研发小组(计算机研发或isRD),负责将尖端技术调整和集成到我们的技术堆栈中,还有一个运营小组(分子架构师或MAs),他们将这些技术应用于与化学团队合作的发现项目和我们的合作伙伴。MA的概念(如图1所示)是融合药物和计算化学的经验和专业知识,在数据科学和计算机工具的基础上工作。我们认为它是建立信任、实现雄心勃勃的目标和加快潜在候选药物发现的有力推动者。MA确保(i)使用正确的工具和方法,无论其来源如何,也无论它们是否使用AI/ML,(ii)数据干净且易于理解,(iii)项目目标明确且得到满足,以及(iv)创建定制的计算管道与高效的可操作DMTA工作流程相结合,以最少的化合物数量测试给定的设计假设。
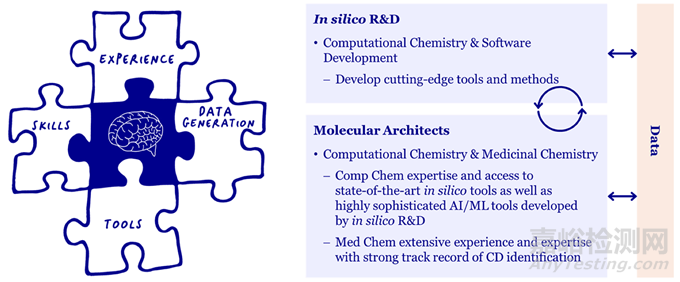
图1 Evotec分子设计的卓越秘诀
D2MTL(Design-Decide-Make-Test-Learn)的概念是由MA引入的,作为成熟的DMTA循环的演变。“Decide”的加入强调了选择阶段与主动学习相结合的重要性,此时优先考虑由人类和机器组合生成的分子设计。我们建议在工作流程中建立高效的Decide阶段是实现卓越分子设计和确保项目顺利进行的关键。这种合作活动不仅为药物优化过程提供了结构,还有助于我们的团队建立对药物和计算化学更深入的相互理解。将“Analyze”替换为“Learn”表征药物化学家的学习以及预测模型的评估和再训练。这种新的工作方式需要与快速综合和测试进行高度集成,以提高循环效率。图2说明了这个概念,并显示了本文中描述的应用程序如何适应。
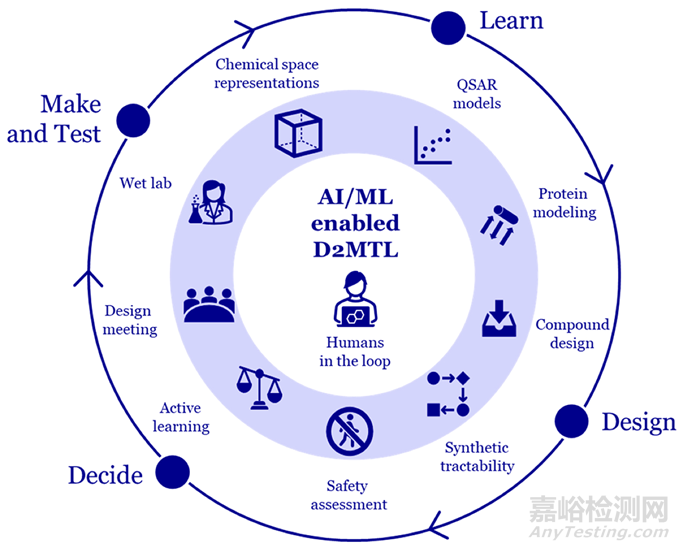
图2 AI/ML技术如何融入设计-决策-制造-测试-学习(D2MTL)循环
结论与展望
人工智能与药物化学的整合是近年来计算化学方法学最重要的发展。预测化合物特性、生成满足特定项目需求的创新设计、确定3D蛋白质结构重排或虚拟筛选数十亿种化合物的能力都是非常有用的发展。我们投资于工具的开发,通常严重依赖开源软件和公开可用的数据,例如经过训练的模型。我们非常感谢AZ等作者和组织,以及拜耳、谷歌DeepMind和麻省理工学院他们决定与药物发现界分享他们的进展。有效使用这些工具需要实验和AI数据科学家、程序员以及计算和药物化学家之间的密切合作。此外,如果我们能够在设计阶段使用AI/ML准确预测新分子的临床前和人体药代动力学特征,则可以显着减少对动物试验的需求。
我们已经介绍了AI对我们的药物化学项目产生积极影响的一些领域。我们预计它的使用会随着对单个技术优点和缺点的认识而增长。然而,由于多种原因,这些方法的采用远非普遍。
例如,生成方法仍然可以产生化学结构,这些结构可能是不稳定的、合成上不可行或非原创的。尽管AI/ML出现了,但几十年来一直困扰QSAR领域的问题仍然存在,例如使用稀疏训练数据对活动悬崖和非累加性进行建模。我们相信,人机协同,或者更准确地说是机器在环,仍然是最好的方法,但这种算法和主观决策的混合意味着相对贡献很难分解。此外,药物化学家抵制制造他们认为有缺陷的化合物,但另一方面,AI生成的设计,即使经过分类和轮次反馈和改进,也并不总是具有足够的质量,以至于它们可以不加检查或未经修改地使用。
仍有许多改进的机会,例如创建更符合特定项目需求的潜在空间、在生成式设计工具中增加3D组件的使用、改进AI的可解释性以及使用内置合成路线创建的生成式设计。后两个方面反过来应该会提高计算经验不足的化学家的采用和文化接受度。最近在深度学习架构中纳入了基于物理的方法,这使得计算属性、优化分子几何形状和分析扭转角成为可能,量子力学(QM)精度大大降低了计算成本。然而,在科技巨头的世界里,重点一直是使用深度神经网络做所有事情。例如,Google DeepMind和Isomorphic Laboratories开发了AF3不使用AF2中采用的力场建模。由于数据的稀缺性和分子环境的复杂性,我们预计物理学、化学和数据科学仍然是必需的,并且可以通过更大的整合来增强它们。在可预见的未来,我们还期待人工智能帮助从化学文献中提取数据,预测蛋白质-配体复合物的构象选择,并提供经过药物化学训练的基于LLM的虚拟助手。
这不是AI在某种人机竞争中超过人类水平表现的问题。几十年来,一些药物化学家一直使用计算工具来辅助他们的工作。这句话归功于DerekLowe,“AI不会是药物化学家的终结,但它将是不使用AI的药物化学家的终结”更接近我们的思维方式。我们不认为这是威胁性的。在1960年代,你可以在那句话中用AI代替QSAR。我们希望通过结合语言(包括SMILES字符串、化学名称、蛋白质序列等)、图像(例如结构草图)、3D结构信息(例如蛋白质-配体复合物)和组学数据(例如转录组学)来变得更加强大。我们相信,在可预见的未来,像我们这样的组织有能力继续从AI/ML的发展中受益。
我们介绍了我们在工业药物化学中使用AI/ML的观点和经验。尽管我们对这个话题充满热情,但我们还是试图从我们的描述中去除炒作,并带来现实的视角。与技术本身同样重要的是采用的组织、沟通和文化方面。我们同意Griffen及其同事的观点需要一种不同的工作方式,这应该会在不久的将来带来社会效益。
参考资料:
https://doi.org/10.1021/acs.jmedchem.4c03044

来源:Internet


