您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2025-03-18 08:43
2025年3月17日,国家药品监督管理局(NMPA)发布了《药品生产质量管理规范(2010年修订)》无菌药品附录(征求意见稿),征求意见的截止日期是2025年5月30日。本文以国内发布的征求意见稿为基础,对比和PIC/S无菌附录的异同。
整体对比
对比的异同分为4个结果:
内容一致,表述相同或略有差异:表达的内涵一致;
内容基本一致,详略程度有差异:中国和PIC/S的整体要求一致,但在详略程度上可能有差异;
要求不同:针对同一话题,中国和PIC/S的要求有差异;
中国特殊要求:仅中国要求,而PIC/S未做明确说明;
请注意:本文仅对比无菌附录的相关要求,未拓展至相关指导原则,如NMPA 无菌工艺模拟试验指南(无菌制剂)对APS给出了详细指导。
各章节整体的对比结果如下:
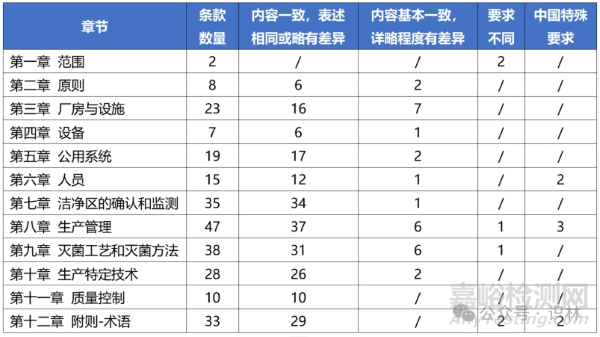
各章节主要差异
第一章 范围
不同要求:征求意见稿本章节未做修改,PIC/S无菌附录适用于所有无菌产品,而中国无菌附录因为有药品的限制,不涵盖辅料、内包材等,征求意见稿在附则中提到,无菌辅料和直接接触药品的无菌包装材料等无菌产品的生产应当参照本附录执行。
第二章 原则:
对应PIC/S无菌附录的第2和第3章,多数条款一致。
内容基本一致,详略程度有差异:本章节中,PIC/S无菌附录对CCS的要素描述更为详细,如PIC/S强调CCS的开发需要详细的技术和工艺知识,具体的CCS要素中,国内用词为纠正措施和预防措施,PIC/S表述为预防机制 - 趋势分析,详细调查,根本原因确定,纠正和预防措施(CAPA)以及对综合调查工具的需求。
第三章 厂房与设施~第五章 公用系统
对应PIC/S无菌附录的第4章~第6章,要求上没有明显不同。
第六章 人员
对应PIC/S无菌附录的第7章。
征求意见稿的特殊要求:1. 无菌药品生产企业的生产管理负责人、质量管理负责人、质量受权人的资质—与《国家药监局关于加强药品上市许可持有人委托生产监督管理工作的公告(2023年第132号)》一致,要求具有至少五年从事药品生产和质量管理的实践经验,其中至少三年无菌药品生产和质量管理的实践经验;2. 中国要求未经培训的外部人员进入洁净区均需要进行详细指导和监督,而PIC/S仅对未经资质确认的人员进入无菌生产区进行了要求。
第七章 洁净区的确认和监测
与PIC/S无菌附录相比,本章节将原属于第4章和第9章中关于洁净级别确认和环境监测的内容整合到了一起,绝大多数条款与PIC/S保持了一致,包括环境监测的标准等,需要注意的一点是,中国要求A级或B级洁净区检测到的微生物应当鉴定到种。
第八章 生产管理
对应PIC/S无菌附录的第8章和第9章,将无菌工艺模拟试验(APS)的内容也整合进来。
不同要求:本章节提出了对污染控制策略(CCS)的一般性要求,保留了原附录中关于无菌药品批次划分、最终灭菌产品的生产操作环境示例的描述,另外需要注意的是,本次征求意见稿对A级送风环境提出了明确要求,即应当至少符合A级区的静态要求;APS方面,征求意见稿中未加入“除非有意模拟厌氧,否则需要用空气代替日常无菌生产工艺所用的任何惰性气体。在这些情况下,应考虑将偶尔的厌氧模拟作为整体验证策略的一部分(参见第9.33段iii)”的描述;中国对灌装数量的描述更细致,生产批量大于10000支时,模拟灌装数量不得低于10000支。
第九章 灭菌工艺和灭菌方法
对应PIC/S无菌附录的第8章。
内容基本一致,详略程度有差异:征求意见稿对于指示剂的使用未采用PIC/S的描述:如果使用了BI,应采取严格的预防措施,以避免将微生物污染转移至生产或其它检验过程;仅BI结果不应代替其它关键参数和工艺设计要素;指示剂仅显示灭菌工艺过程已经发生,并不指示产品的无菌性或指示产品已达到要求的无菌保证水平;
不同要求:在辐射灭菌章节中,PIC/S强调,辐射灭菌主要用于热敏感物料和产品的灭菌。紫外线照射不是可接受的灭菌方法;征求意见稿对辐射灭菌列出了详细要求(保留了原附录的描述),其要求与PIC/S GMP附录12药品生产中电离辐射的应用基本一致。中国还强调:生物指示剂可作为一种附加的监控手段。
第十章 生产特定技术
对应PIC/S无菌附录的第8章,多数条款一致,针对吹灌封技术,征求意见稿中还强调BFS环境监测的静态和动态微生物和悬浮粒子均需满足相应洁净级别的限度要求。
第十一章 质量控制
与PIC/S无菌附录第10章一致。
第十二章 附则-术语:
不同要求:征求意见稿中共33个术语,其中“动态”“静态”的定义均保留了原附录的描述,与PIC/S无菌附录的描述有差异,动态的定义中,中国为规定数量,PIC/S为最大数量人员,对于静态,PIC/S还强调了所有公用设施(包括任何正常运行的HVAC)已安装完成;主要(main)生产设备按规定安装但未运行,而非中国定义中所有生产设备;征求意见稿中还有2个PIC/S中没有的术语:(二十八)无菌生产区:指非最终灭菌产品采用无菌生产工艺的无菌操作所处的区域,该区域通常为A级。无菌生产工艺在本附录中也称作无菌生产;(三十二)药品质量体系(Pharmaceutical Quality System,PQS):指为在质量方面指导和控制制药公司而建立的管理体系。(依据ICH Q10)
整体来看,本次修订中85%的条款与PIC/S无菌附录保持了一致。早在2023年3月,国家局核查中心组织各省局启动与PIC/S GMP对标课题,通过GMP对比得出结论:中国药品GMP与PIC/S的GMP在框架机构、原则理念上基本一致,主要内容基本等同。中国药品GMP在生物制品批签发管理、物料及取样管理的操作要求、对于高致敏性高活性或毒性物质控制等方面要求更详细、更具体。有实质差异的条款,主要集中在无菌药品附录。本次无菌附录修订也是我国申请加入PIC/S的重要工作之一。

来源:识林


