您当前的位置:检测资讯 > 监管召回
嘉峪检测网 2025-03-27 08:52
摘要
美国FDA警告信作为一种重要的监管工具,在美国药品市场中发挥关键作用,旨在监督和确保药品生产企业遵守严格的生产和质量标准,以保障消费者的健康和安全。本文旨在系统介绍美国FDA警告信的起源、法律性质、适用范围和签发程序,探讨了在新形势下推进监管科学创新发展的关键问题以及该制度对促进我国药品监管高水平发展的借鉴意义。
【关键词】警告信;药品监管;合规
美国FDA在日常药品监管工作中如果发现药品生产企业有违规行为,通常会根据问题严重程度采取警告信、进口禁令和取消上市许可等执法行动。其中,警告信是指美国FDA对被检查对象发出的一种劝告性通知,同时在美国FDA网站上向社会公布[1]。警告信通常会指出药品生产企业具体问题和违规行为,并要求接收者采取必要的纠正措施和改进措施,以确保其产品符合美国FDA的法规要求和质量标准。接收到美国FDA警告信的企业或个人通常需要尽快响应并采取纠正措施,否则可能会面临进一步的法律或经济后果,例如产品召回、制造停止或者法律处罚等。因此,美国FDA警告信对于相关企业来说是一种严肃的警告和提醒,需要认真对待并尽快处理其中指出的问题。本文将详细探讨美国FDA警告信制度的法律性质、适用范围及其签发程序,阐述其对我国制药企业的影响和药品监管的借鉴意义。
1.美国FDA警告信制度的法律定位
1.1 美国FDA警告信制度的由来
美国FDA警告信制度的起源可以追溯到美国FDA监管权力的历史背景。美国FDA于1906年根据《联邦食品和药品法》成立,其职责逐步扩展,涵盖了广泛的公众健康和安全责任。1938年,《联邦食品、药品和化妆品法》的出台大大拓展了美国FDA的监管权力和范围,为其监管食品、药品和化妆品提供了全面的法律基础[2]。美国FDA早期的执法策略主要依赖于诉讼和产品扣押。然而,随着其职责范围扩大,采用更灵活和更高效的方法变得非常必要。20世纪中期,美国FDA开始寻求标准化执法实践,警告信逐渐成为一种正式机制,主要目标是创建一种结构化但灵活的方法解决不合规问题[1]。随着时间的推移,警告信制度已成为美国FDA执法策略的基石,反映了美国FDA对基于风险的透明监管流程的承诺,其已成为美国FDA监管科学的工具之一[3]。
1.2 美国FDA警告信的法律性质
美国FDA警告信虽然不是具有法律约束力的文件,但在监管中具有重要意义。警告信是美国FDA的官方信函,用于通知企业在检查期间或通过其他途径发现的违规行为。虽然警告信本身不构成最终的法律行动,但通常预示着更严厉的执法措施,例如产品扣押、禁令或刑事诉讼[4]。
警告信的法律性质植根于正当程序(due process)原则[5]。正当程序原则是美国宪法中一项基本的法律概念,旨在确保所有人都能在法律面前获得公平和公正的待遇。正当程序原则要求在采取不利行动之前,政府必须给予受影响的个人或组织明确通知。美国FDA在警告信中详细列出检查期间发现的违规行为,这一过程确保企业了解其被指控的具体违规事项,并有机会准备回应。
1.3 美国FDA警告信适用范围
美国FDA警告信制度适用于受美国FDA监管的广泛行业,包括药品、医疗器械、食品、化妆品和烟草制品。该制度覆盖与药品生产质量管理规范(good manufacturing practice of medical products,GMP)、标签、营销和分销相关的违规行为,具体适用领域包括[6]:①GMP违规行为。涉及药品、生物制品等生产和质量控制流程的问题。②错误标识和标签违规。产品标签不准确、误导或不完整,包括缺乏科学证据支持或不符合监管标准的声明。③产品掺假。产品中可能存在有健康风险的污染物或杂质,涉及使用未经批准的成分或未遵循正确的生产规程,可能发生在生产过程的任何阶段。④营销和促销违规行为。误导性广告或未经批准的促销宣传,通常涉及对产品功效或安全性做出没有足够证据支持的声明。⑤进口违规。不遵守进口美国FDA监管产品的有关规定,包括未能满足标签要求、提交虚假信息或未能提供适当的文件。
除此之外,美国FDA警告信制度还涉及与临床试验、上市后监测和不良事件报告相关的违规行为。广泛的适用范围确保美国FDA能够有效地监控和执行其监管职责,保障各个方面的合规性。
2.美国FDA警告信的内容及签发程序
2.1 美国FDA警告信流程概述
警告信的流程一般始于相关企业接受美国FDA检查。检查完成后,检查员将发现的偏差和问题以483表格的形式提交给企业,并将现场检查报告(establishment inspection report,EIR)提交给美国FDA办公室进行审查。审查结束后,EIR会最终转发给企业。如果EIR涉及强制采取整改措施(official action indicated,OAI)的要求,则会触发警告信[7]。
首先,美国FDA对企业的检查通常包括对生产设施、质量控制程序和产品合规性的全面评估。检查员根据检查中发现的问题编写检查意见,记录在483表格中并提交给企业。483表格列出了检查中发现的所有偏差或问题,企业管理层需要在规定时间内作出回应,解释如何纠正这些问题。如果483回应不能让美国FDA满意,则可能触发美国FDA警告信。
之后,检查员将根据检查结果和企业的483回应编写EIR。EIR是对检查过程的详细记录,包含所有发现的偏差、企业的回应以及检查员的建议。EIR提交给美国FDA办公室进行审查,确保检查结果和对企业整改计划的期望符合联邦法规的要求。如果EIR涉及OAI的要求,则标识为官方行动指示(OAI)。OAI表示企业存在严重的合规问题,需要采取重大措施予以纠正。如果企业的回应和整改计划未能在规定时间内达到美国FDA的要求,美国FDA将发出警告信。
此外,如美国FDA针对特殊事件(投诉、广告宣传等)的调查或美国FDA实验室检验等其他情形也可能触发警告信。警告信发布流程见图1。
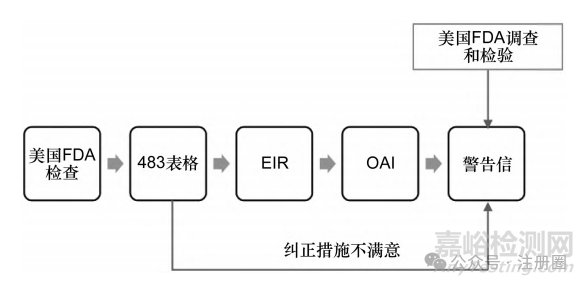
▲图1-美国FDA警告信流程图
2.2 警告信的主体内容
动态药品生产管理规范(current good manufacture practice,cGMP)检查或临床试验现场检查后收到的警告信通常遵循特定格式。美国FDA警告信通常包括以下关键部分:①检查概要。介绍总结了检查的具体情况,例如检查地点的地址和发出警告信的检查日期。②违规识别。对观察到的违规行为进行详细描述,并引用已违反的具体监管要求或法规。③检查结果。包括美国FDA检查结果摘要、相关观察结果和收集的证据。警告信中列出了检查中发现的违规行为、企业对483表格中有关违规行为回应的简要说明以及美国FDA认为回应不充分的原因。④纠正措施。针对企业必须采取的纠正措施提出建议或要求,以解决已发现的违规行为。本节通常包括实施纠正措施的具体时间表。⑤回复要求。指示企业在指定时间范围内(通常为15个工作日)做出回复,详细说明已采取或计划采取的纠正违规行为的步骤。回复必须全面,并解决警告信中确定的每项违规行为。⑥潜在后果。警告如果违规行为未得到充分解决将可能采取的执法行动。
2.3 警告信与483表格的关系
483表格的正式名称为“检查观察”,是美国FDA检查员在检查结束时签发的重要文件,列出了检查员对可能违反《联邦食品、药品和化妆品法》或相关法规的观察结果,其可作为签发警告信的基础。483表格和警告信之间的关系是顺序性和程序性的:①检查和483表格签发。在美国FDA检查期间,检查员在483表格上记录观察结果,并在检查结束时与企业管理层讨论这些结果,讨论为企业提供了一个提出问题的机会并寻求对观察结果的澄清。②评估和决定。美国FDA评估483表格的观察结果,以确定是否需要发出警告信。考虑因素包括违规的严重程度、对公众健康的潜在影响以及企业的合规历史。评估过程可能涉及多位美国FDA办公室和法律人员的意见。③发出警告信。如果美国FDA决定发出警告信,通常会根据483表格中记录的观察结果提供更多详细信息和背景信息。警告信还可能包括其他来源的调查结果,例如不良事件报告或消费者投诉。
2.4 警告信的签发程序
美国FDA警告信的签发程序主要包括以下几个步骤:①检查和记录。美国FDA检查员对受监管的设施进行例行或有针对性的检查,如果发现潜在违规行为,则将观察结果记录在483表格中。检查员还可以收集样本、拍照和进行访谈以支持其发现。②内部审查和评估。美国FDA合规官和法律人员会审查483表格中的观察结果,以评估其重要性并确定适当的监管回应。审查过程包括评估对公众健康的潜在影响和企业的合规历史。③起草警告信。如果需要发出警告信,美国FDA工作人员将起草警告信,经过多轮审查和修订,以确保准确性和清晰度。④签发和交付。最终警告信由美国FDA高级官员批准并签发给企业。该信通常通过挂号信发送,以确保企业收到。⑤回应和跟进。企业必须在规定时间内回复警告信,详细说明已采取或计划采取的纠正措施。美国FDA将审查回应,并可能进行后续检查,以验证纠正措施的实施情况和有效性。
这一结构化流程确保美国FDA警告信是基于彻底的检查、严格的内部审查和对监管要求的清晰传达,有助于维护美国FDA执法行动的完整性和可信度。
2.5 警告信的关闭程序
为确保已发现的违规行为得到充分解决,美国FDA会采取一系列步骤完成警告信的关闭程序,确保美国FDA的执法行动能够持续合规并改善公众健康和安全,其主要涉及以下几个步骤:①纠正措施审查。美国FDA审查企业对警告信的回复,包括已采取纠正措施的文件,评估过程包括审查企业的政策、程序和记录,以确保纠正措施有效且可持续。②后续检查。如有必要,美国FDA将进行后续检查,以确认纠正措施的实施情况和有效性。后续检查可能涉及对企业运营情况的详细审查,包括对员工的访谈和对生产过程的观察。③发出关闭信函。如果美国FDA确定违规行为已得到充分解决,则会发出关闭信函。关闭信函承认企业的合规努力,并正式结案警告信函。关闭信函是一份公开文件,可作为企业已实现合规的证据。关闭信函只能基于企业已实施的纠正措施而签发,而不是仅凭计划签发。根据问题的严重程度,企业可能需要数月到数年的时间才能找到解决方案。如果违规行为无法纠正,则不会发布关闭信函[8]。
3.美国FDA警告信制度的影响和意义
美国FDA警告信制度在药品监管中具有显著意义,其核心在于提升监管透明度和问责制、促进自愿遵守、提供持续改进的框架以及支持执法和威慑。我国药品监管部门也有类似的制度,如告诫信等[9],可以借鉴美国FDA警告信制度的设计理念,持续充实和完善我国相关制度,通过明确告诫行为的制度流程,提升监管透明度,强化自愿合规文化,并增强告诫的威慑作用,进一步提高药品监管效率[10-14]。
3.1 持续加强监管威慑性
警告信通常是采取更严厉执法行动的前兆,例如产品扣押、禁令或刑事起诉,这为企业提供了在面临更严重后果之前自愿纠正违规行为的机会。警告信的威慑作用同样显著,其公开性质和进一步执法行动的可能性为企业提供了强大的合规动力,有助于确保企业优先考虑合规性并主动采取措施避免违规行为。我国的药品告诫、约谈监管方式与美国FDA的警告信制度类似,但大多由地方药品监管部门发出,不同地区企业面临的监管力度不一,建议建立全国统一的告诫、约谈平台,统一监管尺度。此外,告诫行为的公示范围和力度也较弱,未能有效发挥应有的威慑作用。建议参考美国FDA警告信的形式和签发程序,特别是通过责令告知通知书向社会公开,以增强其惩戒性和威慑性。
3.2 促进企业合规自律
美国FDA警告信制度的核心目标之一是促进受监管企业自愿遵守相关规定,通过详细说明观察到的违规行为及所需纠正措施,警告信鼓励企业主动行动以解决不合规问题。这种方法符合美国FDA的监管理念,即通过教育和合作而非单纯依赖惩罚措施监管。自愿合规在制药行业尤为关键,因为药品生产流程复杂,对公众健康的潜在影响巨大,需要高度的监管监督和行业自律。通过培养合规文化,美国FDA帮助企业将公众健康和安全置于优先地位,这对我国药品监管具有重要的借鉴意义。我国2021年新修订的《中华人民共和国行政处罚法》第三十三条新增“首次违法可不处罚”制度,体现了“处罚与教育相结合”的原则。在药品监管领域,可以通过警告信这一告诫行为明确“首违不罚”原则,并结合企业诚信名单等行政工具,进一步提高告诫行为在药品监管中的地位。
3.3 增强监管透明度
美国FDA警告信制度显著提升了监管流程的透明度。警告信作为公开文件,通常会伴随新闻稿和其他形式公开发布,这种透明度使公众和其他利益相关者能够了解美国FDA的执法行动及其背后的原因。警告信的公开性质还具有阻止违规行为的作用,使企业意识到不遵守监管要求可能引发公众审查,进而损害声誉。这促使企业优先考虑合规性,并在发现违规行为时迅速采取纠正措施。相比之下,我国药品监管部门的检查通报在形式上与美国FDA警告信有相似之处,但在具体内容和公开程度上存在差异。为了提高透明度,我国在未来的检查通报中可详细列明违规行为的具体情形及违反的法律规范条款,以便企业能够快速识别问题并采取纠正措施。
3.4 推动监管科学建设
监管科学旨在通过开发和应用科学方法评估药品的安全、有效和质量,从而提高监管效率并推动创新。在这一背景下,美国FDA的警告信制度显得尤为重要。警告信制度为美国FDA提供了大量关于企业合规状况的数据,使其能够分析行业趋势,识别常见违规类型,并制定更有针对性的监管政策。我国药品监管部门可参考这一制度,通过建立类似的警告信机制,提高监管的效率和透明度。警告信制度将积累大量企业合规数据,有助于我国药品监管部门分析行业趋势,识别常见问题,从而制定更科学、更有针对性的监管政策,提升监管效果。
参考文献
[1]宋华琳,刘炫.美国 FDA 警告信的制度架构及启示[1].中国食品药品监管,2019,12:28-35.
[2]US Food and Dng Adminisimtion, About the FDA : History[ EB,OL](2018)[2024-07-18].hilps://www. hh. sov/aboutRhwfia-hiskory.
[3]US Food and Drug Administralion, Advanwing regulatory seienee[EB/0L].(2022)[2024-07 -18].hups ://www. kla. sov/sciece-mseml/sciemeamsearch-specia-lopics/adlwaneingregulaloty-scien!e.
[4]US Food and Dmg Administmtion. lepedtion basies[EB/0L].(2024)[2024-07-18],hilps://www,hha. sowinspedtionscanplianeoralcnmil-inwsliga is'inpecion-ssics.
[5]US Food anl Drg Adinisimtion, Cadle of Feckral Regulalions.(n.d.). Tile2l,Volum:2, Pat 1l5, Sedion 115.50[EB/0L].[2024-07-18].hips://www. accessdlata. &hn. sov/seniplsedveRkeselCFRSmmh, c? f=115.50.
[6]US Food and Drug Administmtion. Regulatory prcedures mama![EB/0L].(2024)[2024-07 -18] hps://www. in. sw/in.specho-onpliae-nonent-acrinina-ivesligalons/compliane-mmsregulaloypmelures-mml.
[7]SAINI CD, MIGLANI A, MUSYUNI P,et al. Review of fonn483s anl warning leters lo plmceulical mau bturers issuedby UsFDA[」]. juma! Generie Melicires,2022,18(1): 32 -41.
[8]US Food an Drug Adminislmlion, About waming anl close-ouleles[EB/0L],(2024)[2024-07 -18]. hps://www. ha.gov/irspeelonssomplianee-enlorcement ancnminal.invesligg-lot"waning ldler"al waming nl cl ml-eter
[9]程立、美国 FDA 警告信制度对我国药品监管工作的启示[J].行政科学论坛,2022.9(8):47 -50.
[10]陈钊锃,夏逸凡,李慧、2016 至2020 年美国FDA 对中国,印度制药企业所发警告信的综合分析[11.中国医药工业架点,2023,54(9):1391-1395.
[11]曹琳琳,钱雅婷,张青松,等、FDA 发给中国药品生产企业eGMP警告信的分析与启示[J],中国医药工业杂志,2023,54(6):950-956.
[12]李武超,马岩松,原茵,等,生物制品生产质量风险管理常见问题分析与建议[J].中国医药导刊,2024,26(4):359-34
[13]王波,邵蓉.FDA 指导原则管理规范的发展及启示[].中国现代应用药学,2023.40(14):2010-2015.
[14]刘艺迪,何辉,周刚,美国 FDA 药物临床试验研发主体合规检查信息公开情况的介绍和启示[1].中国新药杂志,2024,33(2):124-130.

来源:中国新药杂志


