您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-04-07 08:29
目前在研的免疫调节药物数量逐日剧增,形式也丰富多样,比如单抗、双抗、基因修饰T细胞、免疫-药物偶联物、基因治疗等等。所有这些药物多多少均涉及到免疫毒性评价。
众所周知,ICH S8是主要介绍人用药物免疫毒性研究的指导原则。不过,该指导原则颁布距今18年,已经不能完全满足工业界的需求。ICH S8于2006年颁布时,工业界开发的免疫药物主要为免疫抑制方向,药物形式也以小分子化药为主。时过境迁,现如今是个百花齐放的时代,生物药物爆发式增长,肿瘤免疫治疗已经成为时代新宠,其它细胞和基因治疗药物的研发也如火如荼。这些新的药物形式是不是适用于ICH S8,如果适用,需要开展哪些研究,都需要指南给出更具体的建议。
2023年,FDA出台了新的免疫毒性研究指南(Nonclinical Evaluation of the Immunotoxic Potential of Pharmaceuticals)。不过,依然未能解决新分子形式药物免疫毒性研究需要开展哪些测试以及监管机构接受的终点是什么。
Pfizer、Seagen、Abbvie、Boehinger-Ingelheim、Takeda、Gilead、GSK等组成的Immunosafety Working Group of IQ/DruSafe对工业界免疫毒性研究的挑战进行了一组调查,分享如下。
共16家药企给予了反馈,其中13家是雇员超过万人的企业,3家雇员1000-10000人企业。所有企业均一致认为免疫安全性研究是非临床开发中的关键组成部分。开展免疫安全性研究的主要原因分为3类:1)患者安全性;2)评估在靶和脱靶作用;3)加深对作用机制的理解。
治疗领域方面,免疫学/炎症领域,100%药企均会开展免疫毒性评价。肿瘤免疫治疗领域,这一比例是15/16。非免疫治疗抗肿瘤领域和抗感染领域这一比例就低很多,分别是8/16、3/16。
大部分企业(15或16/16)会在非临床研究中加入以下毒理终点,用于免疫安全性评估,包括临床症状、组织病理学、血液学、常规免疫表型、CRA(cytokine release assay)、血清细胞因子/趋化因子、T细胞亚群(活化T和记忆性T)、TDAR(T-cell-dependent antibody response,T细胞依赖抗体反应)。还有很多企业会开展额外的ex vivo研究。这些指标已经超过ICH S8中的常规毒性研究内容,包含了附加的免疫毒性研究。ICH S8和CDE于2024年1月最新出台的《药物免疫毒性非临床研究技术指导原则》均指出,如果常规毒性研究提示存在免疫毒性,应根据观察到的免疫学改变的性质和化合物类别相关的担忧决定采用何种适用的附加免疫毒性试验方法。推荐进行免疫功能研究,如TDAR。
缺少合适的临床前模型、缺少合适的检测方法、监管机构接受标准的不确定性、ADA结果的可转化性是目前非临床免疫毒性评价面临的主要挑战,其中缺少合适的动物模型排名最为靠前。关于这点,可能的原因有很多,比如靶点仅在人体表达,种属间的靶点序列或结构相差太多。还有些靶点仅在疾病状态下表达,而非临床研究主要采用健康动物开展。比如CD28激动剂TGN1412,猴中的耐受性良好,临床则出现严重的细胞因子风暴,与CD28靶点在动物和人体中的表达差异有关,导致非临床向临床转化出现问题。
重点介绍下TDAR和CRA。
TDAR
有些朋友可能对TDAR这个概念还不熟悉。TDAR主要用于免疫系统整体功能性评估。通常是对动物注射高免疫原性的蛋白如KLH,检测抗KLH抗体,判断机体免疫状况。从免疫原识别到抗体产生,体内经历了巨噬细胞/DC细胞和/或B细胞的抗原递呈功能、辅助T淋巴细胞激活、B淋巴细胞抗体产生等一系列免疫过程,是检测免疫抑制作用的重要方法之一,也可用于检测免疫增强作用。TDAR可单独开展,也可伴随在重复给药毒性试验中考察。
14/16企业反馈会把TDAR包含在非临床研究中。传统认为TDAR主要用于检测免疫抑制,不过已经越来越多用于免疫激活或免疫增强作用的评估。TDAR数据对于解释药物的药理学和免疫毒理学,及临床风险转化方面非常有价值。
那么如何对TDAR的adversity进行定性呢?10/14企业是将TDAR出现的供试品相关的改变和组织病理学检查结果作为确定adversity的主要依据。也有很多企业将TDAR改变与血液学和免疫表型之间的相关性进行分析,作为判定adversity的依据之一。其它考量因素还包括临床症状、ex vivo功能性试验终点、总的证据权重分析等。TDAR结果可以用于提示临床风险。比如临床前出现TDAR结果异常,临床方案中安全性需监测感染风险、考虑对疫苗接种的影响等。
TDAR通常不单独用于在毒理学研究中确定NOAEL。即使TDAR结果变化幅度显著,但其它毒理结果未见变化,通常也不影响NOAEL的确定。然而,当TDAR的变化幅度与其他参数变化(包括组织病理学、血液学和免疫表型终点)同时存在时,可以基于TDAR的变化幅度进行NOAEL的判断。
CRA
14/15的企业会将CRA包括在免疫毒性评价中。具体到哪些药物形式需要开展CRA研究,如下图所示。占比最高的原因是基于合理性考量,比如靶点表达在免疫细胞表面。其它原因包括新型生物制品、免疫检查点抑制剂、新的生物药物联合、基因工程修饰的T细胞治疗等。国内CDE出台的《药物免疫原性研究技术指导原则》明确提出,对于免疫调节类药物,除了在动物体内检测细胞因子外,还需要开展体外细胞因子释放试验。不过,CDE《药物免疫毒性非临床研究技术指导原则》也有提到,当药物不直接结合到参与免疫系统激活的表面受体时,通常不需进行细胞因子释放试验。关于这点,指导原则没有过多展开,个人理解如果药物靶点不是免疫细胞表面的膜受体,比如是一些游离可溶性靶点,体外CRA试验可以不用开展?

CRA中检测最多的细胞因子是IFNγ(15/15)、TNFα(15/15)和IL-6(14/15)。其次是IL-1β、IL-2、IL-4、IL-8、IL-10、IL-12p70,占比9/15,说明大部分企业会检测以上细胞因子。当然,绝大多数企业(14/15)还会根据供试品的药理学作用,增加特异性细胞因子检测。大部分企业用全血或PBMC进行细胞因子释放的检测。具体检测模式选择固相法还是液相法,没有标准答案,大部分企业会根据靶点是否表达在免疫细胞表面、是不是可溶性靶点、是否需要交联发挥作用、是否表达在内皮细胞等进行决策。国内CDE要求固相法和液相法均尽可能要考虑,检测的细胞因子至少包括IFNγ、TNFα、IL-2、IL-4、IL-6、IL-8、IL-10。建议采用未被刺激的人源细胞、全血和/或其他基质进行评估。
关于CRA结果对于临床的指导意义,首先大部分企业会用CRA的结果进行起始剂量计算,即MABEL法。CRA阳性结果会影响起始剂量安全因子设定及剂量爬升阶段的剂间距设计。其它额外的临床考量点还包括患者预处理、哨兵法设计等。

CRA的挑战在于对结果的解读。企业反馈排名最靠前的是如何解释CRA结果的临床相关性。说的直白点,临床前CRA中细胞因子的升高,临床一定会升高吗?临床前不升高,临床一定不升高吗?这中间的转化效果和指导意义尚没有统一的判断标准。其次是CRA中细胞因子升高到什么浓度才会引起担忧。不同机制免疫调节剂的免疫刺激作用是不同的,CD28激动型抗体明显强于细胞因子,细胞因子大概率也比免疫检查点抑制剂强。但很难判定具体某一因子升高到某一浓度,会引起比较大的担忧。其它难点还包括如何分析数据(看趋势还是进行统计分析)、如何选择合适的阳性和阴性对照、联合用药如何考量、应该检测哪些细胞因子/趋化因子等。
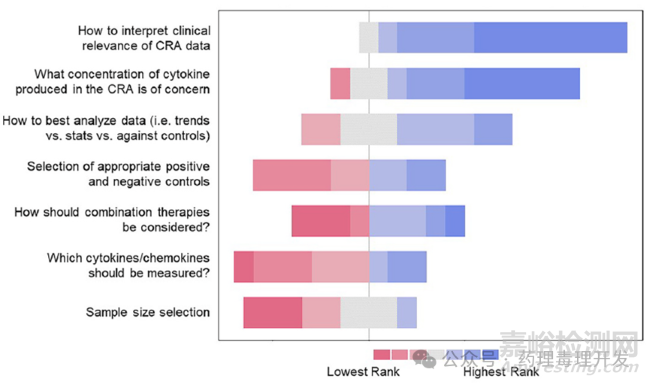
目前的CRA通常不用于对风险进行定量评估,因为缺乏数据分析的标准方法以及对其临床相关性的理解。其他原因还包括治疗靶点可能在健康志愿者的细胞中缺失,疾病状态下免疫细胞的状态可能不同,靶点表达水平在个体之间存在差异,而且这些检测可能缺乏与其他生理系统的关键相互作用(在模拟整个生物体反应方面的局限性)。此外,患者血清或血浆中观察到的细胞因子变化可能与体外CRA中观察到的细胞因子变化不直接相关。虽然监管机构期望进行CRA,并且一旦发现阳性结果,则应在临床研究中采取更谨慎的方法(如低起始剂量、缓慢增加剂量等),但其真正的非临床预测价值尚不清楚。

来源:药理毒理开发


