您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-11-21 15:50
采用扫描电子显微镜(SEM)与X射线能谱仪(EDS)联合应用的方法对材料进行微区化学成分分析是一种重要的分析手段,该方法方便快捷,对不平试样也可以用无标样定量程序得到较好的定性、半定量分析结果,应用较为广泛。
1、 EDS定量分析的基本原理
EDS是一种能采集X射线、测量X射线强度,并计算其与X射线能量函数关系的设备。EDS利用SEM探测器,通过检测电子与试样相互作用产生的特征X射线波长对试样进行定性分析,再根据X射线的强度对试样进行定量分析。定量分析的原理为:X射线能谱仪试样中元素A的相对含量与该元素的特征X射线的强度IA(X射线计数率)成正比,计算方法如式(1)所示。

定量分析分为有标定量分析和无标定量分析。无标定量的分析结果主要是通过理论计算标样的特征X射线强度或者数据库处理获得的,无需测量标样。一般而言,常用EDS能检测到的元素质量分数下限为0.1%。元素分析范围为Be元素到U元素,原子序数为4~92。
2、 EDS在定量分析中的应用
SEM具有放大倍数大、分辨率高、景深大等特点,结合X射线能谱仪可自动、快速地对元素组成进行定性或半定量分析。EDS定量分析的方法一般分为点分析、线扫描分析和面扫描分析,3种分析方法各有特点,根据试样和需求不同可以选择不同的分析方法。
点分析是将电子束固定在试样感兴趣的点上,然后进行定性或定量分析。特征X射线谱线强度与相关元素含量和化学成分有关,且特征X射线作用于试样时会产生一定的深度和侧向扩展( 微米级),因此EDS获得的结果是该体积内的平均值。实际操作中,通常在该试样观察区域的3个点处进行依次采样。一般采用该定量分析方法对材料晶界、夹杂、析出相、沉淀物、材料的组成等进行分析。
线扫描分析是指电子束沿选定直线进行扫描的同时,将对应的信号强度显示出来,该方法可以更直观地表明元素质量分数不均匀性与试样组织之间的关系。因此线扫描分析可用于测定某元素在相区或界面上的富集和贫化现象。另外,线扫描分析可用来测定镀层的分布情况,此时要考虑镀层的厚度,必要时采用试样旋转的方式对试样截面进行观察。
面扫描分析(mapping)是指选择试样的某块区域,在试样表面移动电子束进行往返扫描。特征X射线的强度可以用亮度或色彩来表示。当用亮度表示时,每采集一个点的特征X射线光子,就会在显示器对应位置上打一个亮点,这些亮点就是对元素的映射。mapping中越亮的地方元素含量越高。当用色彩来表示时,mapping中信号较强的地方元素含量越高,因此可用于观察试样中元素分布的不均匀性。图1为一个花岗岩试样断面组织的面扫描结果。

以上3种分析方法各有特点,根据试样的特点、分析目的、经验等合理选择分析方法。如果想直接观察到杂质分布或相分布,可以采用面扫描分析方法;如果需要对试样表面较小区域进行快速扫描,可采用点分析方法。
EDS定量分析常用于材料成分分析、矿物检测、试样微区分析、失效分析等领域。谢金宏利用EDS研究了不锈钢荒管开裂的原因。姜浩等利用EDS对一次性聚丙烯餐盒的添加器、助剂等安全性进行分析研究。采用EDS进行显微成分分析,当选择不同试验条件时,试验结果具有一定的差异。因此,扫描电镜参数、试样处理方式等因素均会影响分析结果的准确性。
3、 提高定量分析结果准确性的方法
能谱法定量分析的误差包含系统误差和分析误差。系统误差主要源于X射线光子计数的统计涨落、计算方法产生的误差,而仪器的稳定性、分析条件等因素会影响结果的分析误差。
3.1 荷电情况
如果试样有严重的荷电情况(见图2),则会影响其形貌观察及化学成分分析结果,此时必须选用相对较小束流进行分析,否则在电子束的轰击下将产生电子束不稳定、图像模糊等问题,无法进行EDS测试。当被分析元素含量接近探测限时,束流较小,因此无法检测到微量元素。必要时需对荷电试样进行镀膜处理。

3.2 镀膜试样
如试样为不导电试样,则需对其进行镀膜处理,处理时必须保证镀膜厚度不大于20nm,不宜过厚,否则会影响试样的化学成分分析结果。高学平等在对有银涂层的SiO2基体进行EDS分析时,发现试样基体表面逸出的X射线受到涂层厚度的影响不会逸出,因此无法分析涂层下面基体的化学成分。
3.3 仪器参数的影响
配置合适的加速电压。在过压比为2~3时,X射线荧光的产额最高,因此分析轻元素用低加速电压,分析重元素用高加速电压。如果试样完全未知,可首先选择20kV作为初步定量分析电压,随后根据试样的化学成分来调整加速电压。如果用5kV电压来分析Cu合金中的Cu元素,则会得到Cu元素的质量分数为0。因为在正常程序下,分析程序会自动选择元素线系。Cu首先选择的是Cu Kα线系,而Cu Kα线系的临界激发电压是8.05kV,5kV电压无法激发Cu Kα。
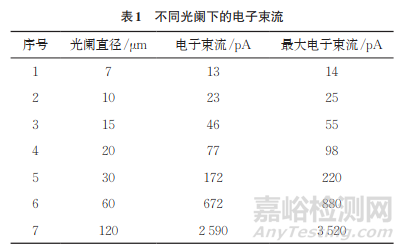
检查束流的大小和稳定性。入射电子束电流简称束流,是通过聚光镜励磁电流调节,强励磁电流的束流小。改变束流的目的是得到合适的计数率。束流越大,能谱仪的计数率越高,但图像分辨率会降低,且损伤试样。信噪比低,对试样表面的损伤小。选择合适的计数率,在不损伤试样和图像质量的前提下,尽量增大束流,使能谱仪的死时间达到最佳的30%~40%。同时确保束流的稳定性。表1为不同光阑下电子束流,光阑越大,束流越大。测试时选择合适的光阑即选择了合适的束流。
选择合适的工作距离。工作距离是物镜极靴下与试样表面之间的距离,是由试样的形状和检出角的几何关系确定的。试样只有在合适的距离下才能采集到高的信号强度。试样安装后,先调节电镜工作距离,使屏幕显示电镜厂家推荐的工作距离合适,然后调节电镜聚焦,使图片清晰。
3.4 计数总量的影响
合理选择加速电压、束流、活时间等条件,尽量保证EDS分析时总计数量达到25万左右。延长收集时间、补充收集效率、增大束流、调整试样的倾斜等操作均会影响计数总量。正常情况下,需保证电子束垂直入射到试样表面,因为试样不平整会使X射线的吸收增加,X射线信号减弱。还要保证电子束垂直入射到试样相对平整的一面。
3.5 合理使用系统软件
以INCA型EDS定量分析系统为例,可以在收谱过程中自动识别并标注元素的峰位,并与标准试样谱库进行拟合。同时,采用自动相位图可以分析多相试样,得到试样的相分布比例图像,提高了对混合相的分析效率。某些软件中的可视化谱峰剥离功能直观有效,可以计算出添加元素的拟合峰,通过比较拟合峰与实际采集的谱峰,判断出试样的实际成分。面扫描分析时,若衬底中的元素与试样中的元素存在特征X射线谱峰重叠现象,会导致谱图中元素的亮点分布异常,无法获得理想的能谱分析结果。此时合理使用系统软件可以有效甄别元素谱图。在矿物的定量分析中,采用集成计算机图形和数据处理软件的物相定量分析系统,可以对特殊试样的采集面积、背景灰度等进行定量分析。
4、 EDS测试时几种常见问题
4.1 检测到试样中原本没有的元素
如果检测到原本没有的C、O等元素,这主要是由于试样的制样、放置等过程中与空气接触,吸附了空气中一些低沸点的挥发性有机化合物。若吸附量达到了X射线能谱仪的检测下限,就能被探测器检测到。如果检测到了一些实际没有的金属物质,如铝、铜等,原因是试样太薄了,电子束流有一定的穿透性。测试含硫物质时,如果出现Mo元素,测试含铟物质时,出现K元素,测试含溴物质时,出现Al元素,原因是出现了重叠峰现象。元素的某些线系间临界激发能差别较小,产生了重叠峰,因此需要对这些峰进行逐一辨别。遇到重叠峰时,解决方法是增大加速电压,激发某些元素在其他线系的能谱峰。如元素Al(K1.49)和元素Br(L1.48)接近,可以增大电压至30kV,通过Br(K11.9)的能谱峰判定是否有元素Br。
4.2 轻元素试样的分析
在测试轻元素时,如C、O、N等元素,这些元素的X射线波长过长,能量较低,使得X射线的吸收增大,因此很难检测到这些元素,或存在很大的不确定性。可以采用延长采集时间、降低加速电压(元素临界激发能的2~3倍)、使用大孔径光阑等方法。试样中通常含有多种元素,若为了增大轻元素的X射线强度,而采用较低的电压,会使试样表面氧化层的元素含量增加。需要注意的是,此时氧元素的质量分数并不代表试样中氧元素的实际含量。
4.3 不稳定试样的分析
在实际应用中,试样分析区域可能出现溶化、开裂、起泡、变黑等缺陷,这样的试样一般称为不稳定试样。由于材料的敏感性不同,电子束轰击时可能会出现质量损失、离子迁移、颜色改变等不同程度的损伤。因此对不稳定试样进行EDS分析时,根据试样特点可选择采用较低的加速电压(如10~15kV)、采用较大的电子束直径和小束流,以及缩短采集时间等方法。
4.4 辨别和避免和峰的出现
和峰是因两个特征X射线光电子同时进入探测器无法分辨而产生两个特征X射线光子能量之和的假峰,和峰强度随计数率的增加而增大。和峰出现在元素能谱主峰的高能量侧,其能量为主峰的整数倍。为了区分和峰,可以采用改变束流、束斑直径等方法减小计数率,与高计数率的EDS进行对比,即可识别某一和峰。
4.5 出峰慢或不出峰问题
在进行EDS分析时,有时候会遇到出峰很慢或无法出峰的情况,可能与死时间过长或输入计数率过低有关。输入计数率会直接影响死时间的长短,计数率越高,死时间越长。
5、 结语
在进行EDS元素定量分析时,需要掌握X射线能谱仪定量的原理、软件处理方式、试样的选择方法、采谱时定量分析条件等内容,并结合实际经验进行分析,研究结果可对从事微区定量分析的相关人员提供一定的参考。
作者:曹艳芬1,曹方方2,孔强强1,宋鹏1,辛玲1,李海娥1
单位:1. 济宁市质量计量检验检测研究院;
2.山东省生态环境监测中心
来源:《理化检验-物理分册》2024年第9期

来源:理化检验物理分册


