您当前的位置:检测资讯 > 实验管理
嘉峪检测网 2025-03-19 08:24
1. 背景介绍
蛋白质是生命活动的核心分子,其功能由其三维结构决定。蛋白质的结构可分为四个层次:一级结构(氨基酸序列)、二级结构(α螺旋、β折叠等局部构象)、三级结构(单个多肽链的全局折叠)和四级结构(多亚基的组装)。
例如,酶的催化活性依赖于活性中心的精确三维排布,膜蛋白的信号转导功能与其跨膜结构域的构象变化密切相关。

(图片来源:https://www.rcsb.org/#Category-analyze)
研究蛋白质结构不仅能揭示其作用机制,还为药物设计、疾病治疗和合成生物学提供基础。然而,蛋白质的尺寸通常在纳米级别,传统光学显微镜无法观测,因此需要借助高分辨率技术解析其原子级结构。
X射线晶体学(XRD)和冷冻电子显微镜(Cryo-EM)是当前两种最主流的蛋白质结构解析技术。
2. 方法学比较
● X射线晶体学(XRD)
X射线晶体学的发展始于20世纪初,英国物理学家William Henry Bragg与其子William Lawrence Bragg开创了X射线晶体学领域。他们提出的布拉格定律揭示了X射线与晶体原子排列的定量关系,为物质结构分析奠定了数学基础。
这项技术的影响之深远,正如奥地利化学家马克斯·佩鲁茨所言:“27项诺贝尔奖不可或缺的配方”——截至20世纪末,X射线晶体学直接推动了27项诺贝尔奖的诞生。1953年,英国科学家罗莎琳德·富兰克林(Rosalind Franklin)利用X射线晶体学拍摄了DNA的B型衍射图(“照片51号”)。
图中的“X”形衍射斑直接提示了螺旋结构的对称性——每圈10个碱基对,螺距34 Å。尽管富兰克林因早逝未能获得诺贝尔奖,但她的这份图像帮助詹姆斯·沃森和弗朗西斯·克里克创建了DNA 分子的双螺旋结构模型,成为了生物学发展的一座里程碑。
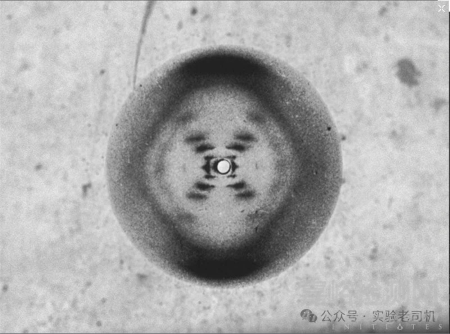
(“照片51号”;图片来源:https://assassinscreed.fandom.com/zh/wiki)
1958年,科学家在首个成像蛋白质—肌红蛋白内发现了不规则的褶皱,首次利用X射线晶体学解析了其结构,标志着X射线晶体学在蛋白质结构分析领域的重大突破。
该技术的核心原理是利用X射线与蛋白质晶体中原子的相互作用:当X射线穿过高度有序的晶体时,晶格中的原子会使X射线发生衍射,形成特定的衍射图案。
通过分析这些衍射点的位置和强度,结合数学方法(如傅里叶变换),可计算出蛋白质的三维电子密度图,进而构建原子模型。
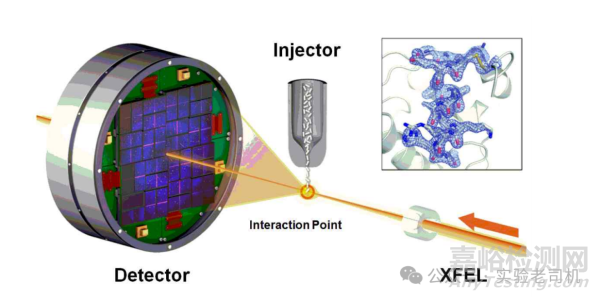
(图片来源:https://images.app.goo.gl/yfVo1LwXBFu5Dsxq5)
然而,该技术面临两大挑战:蛋白质结晶和相位问题。许多蛋白质(尤其是膜蛋白或柔性蛋白)难以形成高质量晶体。例如,G蛋白偶联受体(GPCR)因跨膜区的不稳定性,直到2004年通过引入抗体片段稳定构象才被成功解析。
相位问题则源于X射线衍射仅能记录振幅信息而丢失相位(波的相对位置),需借助分子置换法或引入重金属原子(如硒代甲硫氨酸)来间接解决。
技术的进步显著提升了X射线晶体学的应用范围。同步辐射光源提供了高强度X射线,使数据采集速度更快、分辨率更高。X射线自由电子激光(XFEL)甚至能捕捉飞秒级动态过程,例如光系统II分解水分子时的瞬时中间态。
● 冷冻电子显微镜(Cryo-EM)
冷冻电子显微镜的突破性进展始于2010年代,被称为“分辨率革命”。与X射线晶体学不同,Cryo-EM无需结晶,可直接分析溶液中的蛋白质样品。
其关键步骤包括:将蛋白质样品快速浸入-196℃的液乙烷中,使其在毫秒内冻结成无定形的玻璃态冰,避免冰晶形成(冰晶会挤压样品导致结构畸变)。玻璃态冰中的水分子呈无序状态,包裹生物分子并维持其天然构象。
随后用高能电子束穿透样品并记录散射电子的信号。通过采集数万至数百万张二维投影图像,利用计算机算法(如单颗粒分析)对随机取向的颗粒进行三维重构。
(图片来源:NobelPrize.org)
Cryo-EM技术的核心优势在于解析柔性或超大复合体的能力。例如,核糖体和病毒衣壳(如乙肝病毒)的精细结构均通过Cryo-EM得以揭示。
此外,该技术能捕捉同一蛋白的多种构象状态。在新冠病毒研究中,Cryo-EM解析了刺突蛋白的“开放”(结合宿主受体ACE2)和“闭合”(免疫逃逸)构象,为疫苗设计提供了关键依据。
Cryo-EM技术瓶颈的突破主要依赖于硬件和算法的革新。直接电子探测器(DED)显著提升了图像信噪比,而深度学习算法(如cryoSPARC)实现了高效的数据处理和分类。
目前,Cryo-EM的最佳分辨率已接近2 Å,与X射线晶体学相当,但对样品的纯度、浓度和均一性仍有较高要求。
3. 小结
X射线晶体学和冷冻电子显微镜是研究结构生物学的两大支柱,两种技术并非相互替代,而是形成互补。
X射线晶体学在原子级分辨率上具有优势,以原子精度揭示静态细节,尤其适合小分子量、刚性且可结晶的蛋白质。而Cryo-EM更擅长处理超大复合体(如核孔复合物)或动态过程(如分子伴侣辅助的蛋白质折叠),以动态视角捕捉构象变化。
二者共同推动了蛋白质功能机制的解析。随着技术进步,XRD和Cryo-EM技术正与其他方法深度融合,人工智能(如AlphaFold)的介入进一步加速了结构预测与实验验证的迭代。
这些发展将推动结构生物学从单一分子解析迈向复杂系统的动态研究,最终实现“从原子到细胞”的全尺度生命理解。
4. 参考文献
马礼敦. X射线晶体学的百年辉煌 [J]. 物理学进展, 2014, 34(2): 47–117. doi: 10.13725/j.cnki.pip.2014.02.001
https://digitalpaper.stdaily.com/http_www.kjrb.com/kjrb/images/2014-03/30/02/DefPub2014033002.pdf
Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., ... & Miyano, M. (2000). Crystal structure of rhodopsin: A G protein-coupled receptor. Science, 289(5480), 739-745.
Rasmussen, S. G., Choi, H. J., Rosenbaum, D. M., Kobilka, T. S., Thian, F. S., Edwards, P. C., ... & Kobilka, B. K. (2007). Crystal structure of the human β2 adrenergic G protein-coupled receptor. Nature, 450(7168), 383-387.
Rasmussen, S. G., DeVree, B. T., Zou, Y., Kruse, A. C., Chung, K. Y., Kobilka, T. S., ... & Kobilka, B. K. (2011). Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature, 477(7366), 549-555.
McCoy, A. J. (2007). Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallographica Section D: Biological Crystallography, 63(1), 32-41.
LI Zhifei, GAO Ning. Cryo-EM Method for High-Resolution Structure Determination: A Brief Introduction to 2017 Nobel Prize in Chemistry. University Chemistry[J], 2018, 33(1): 1-6 doi:10.3866/PKU.DXHX201711035

来源:实验老司机


