您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-08-29 19:39
摘 要 · Abstract
药品上市前及上市后的各种变更可能对药品的安全性、有效性、质量可控性产生重大影响,药品变更管理也因此受到各国监管机构和法律体系的严格规定。我国药品变更管理的法律体系在2019 年新修订《药品管理法》及其配套的部门规章与技术指南出台之后开始日趋完善。本文将对我国药品变更管理法律体系的基本框架、历史变迁与最新发展进行概括与梳理,并对进一步完善药品变更管理法律体系提出初步建议。
Drug changes may have major impact on the safety, effectiveness and quality controllability of drugs. Management of drug changes is subject to oversight by government agencies and legal systems around the world. The legal system for drug change management in China has been greatly improved since the promulgation of the newly-revised Drug Administration Law in 2019, its relevant provisions and technical guidelines. This article introduces the basic framework, historical and latest developments of the legal system for drug change management in China, and offers suggestions for further improvement.
关键词 · Key words
药品变更;临床变更;药学变更;药品上市前变更;药品上市后变更
drug changes; clinical changes; pharmaceutical changes; pre-marketing changes; post-marketing changes
01、目前药品变更管理法律体系的基本框架
药品在临床前研究、临床试验、上市许可申请、生产和销售等各个环节都可能发生各种变更。目前规范我国药品监管部门和企业如何妥善管理这些变更的法律框架主要由以下5 部法律规章组成。
《药品管理法》(2019)[1] 建立了总体原则,按照变更对药品安全性、有效性和质量可控性的风险和产生影响的程度,实行分类管理。属于重大变更的,应当经国务院药品监督管理部门批准,其他变更应当按规定备案或者报告。
《药品注册管理办法》(2020)(以下简称《注册办法》)[2] 将变更发生的时间分为药物临床试验期间、药品上市许可申请审评期间和药品上市后三个环节,并作出相应规定。其中上市后变更分为重大、中等或微小变更,分别要求获得批准、进行备案或者提交年度报告。报告类变更途径的新增,在强化持有人责任的同时,也提升了持有人进行变更的及时性和自由度[3]。
《药品上市后变更管理办法(试行)》(2021)(以下简称《变更办法》)[4] 把药品上市后变更细分为注册管理事项变更和生产监管事项变更。注册管理事项变更包括药品注册批准证明文件及其附件载明的技术内容和相应管理信息的变更,具体变更管理要求按照《注册办法》及相关技术指导原则的有关规定执行;生产监管事项变更包括药品生产许可证载明的许可事项变更和登记事项变更,具体变更管理要求按照《注册办法》《药品生产监督管理办法》及《药品生产质量管理规范》的有关规定执行。
《药品生产监督管理办法》(2020)(以下简称《生产办法》)[5] 规定,变更药品生产许可证许可事项的,向原发证机关提出药品生产许可证变更申请;未经批准,不得擅自变更许可事项。变更药品生产许可证登记事项的,应当在市场监督管理部门核准变更或者企业完成变更后30 日内,向原发证机关申请药品生产许可证变更登记。变更生产工艺的,应当按照《药品生产质量管理规范》的要求对生产工艺变更进行管理和控制,并根据核准的生产工艺制定工艺规程。生产工艺变更应当开展研究,并依法获得批准、备案或者进行报告。
《药品生产质量管理规范》(2010)[6] 规定企业应当建立变更控制系统,对所有影响产品质量的变更进行评估和管理。需要经药品监督管理部门批准的变更应当在得到批准后方可实施。《药品生产质量管理规范》对变更的设计、评估及验证,从技术要求的角度作出了很多详细规定。
02、《 药品管理法》实施前药品变更管理的发展
2019 年颁布的《药品管理法》与2020 年颁布的《注册办法》实施以前,我国药品变更法律体系尚未完全成型。法律层面未有药品变更管理的详细规定,药品变更的申请路径与监管要求大多散见于行政规章、通知、公告和指导原则中(图1)。
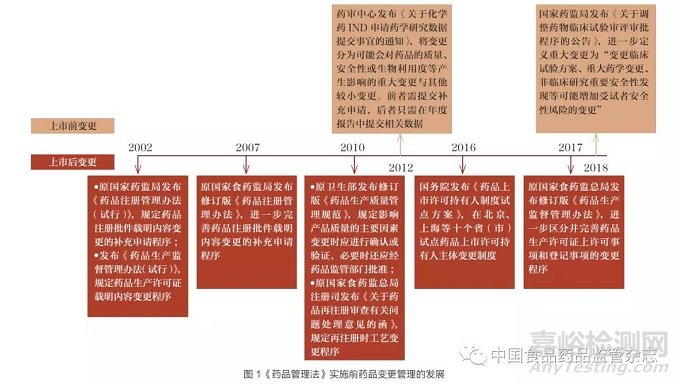
《关于化学药IND 申请药学研究数据提交事宜的通知》[7] 和《关于调整药物临床试验审评审批程序的公告》[8] 等规定虽然弥补了我国上市前变更管理的空白,但这些规定未对临床试验期间变更种类以及评估标准做出详细解释,且未涉及对药物临床试验完成后或审评审批期间变更的规定,当时的药品上市前变更管理制度具有较大的不确定性[9]。
此外,尽管国家药监局药品审评中心(以下简称“药审中心”)陆续发布了《生物制品生产工艺过程变更管理技术指导原则》(2005)[10]、《已上市化学药品变更研究的技术指导原则(一)》(2008)[11]、《已上市化学药品生产工艺变更研究技术指导原则》(2017)、《已上市中药变更研究技术指导原则(一)》(2011)等指南文件,以落实上市后的变更管理,但各技术指导原则中对变更的分类与要求并不统一。例如,《生物制品生产工艺过程变更管理技术指导原则》将变更划分为Ⅰ、Ⅱ、Ⅲ类变更,其中Ⅰ类为引起产品内在质量的改变、需按照新药申报程序进行申报的重大变更;而《已上市化学药品变更研究的技术指导原则(一)》却将Ⅲ类变更作为重大变更,Ⅰ类变更作为微小变更。可见,相关指南文件在起草时缺乏系统化的考量,从而可能造成不统一的管理局面。
由此可见,在《药品管理法》及其配套规定出台以前,我国药品变更管理法规尚缺乏统一性和系统性,对申请人或持有人如何管理变更和国家如何有效监管造成一定困惑。随着新修订《药品管理法》及其配套规定的出台,药品上市前与上市后变更的管理法规日趋完善,形成了行政法律制度为主、技术指导原则为辅的药品变更管理制度体系。
03、《 药品管理法》实施后药品变更管理法律体系的最新发展及改进建议
3.1 药品上市前的变更管理
3.1.1 临床试验期间的变更
《注册办法》首次在规章层面对临床试验期间的药品变更程序进行了规定。根据《注册办法》第二十九条规定[2],药物临床试验期间,发生药物临床试验方案变更、非临床或者药学的变化或者有新发现的,申办者应当按照规定,参照相关技术指导原则,充分评估对受试者安全的影响。申办者评估认为不影响受试者安全的,可以直接实施并在研发期间安全性更新报告中报告。可能增加受试者安全性风险的,应当提出补充申请。药审中心对补充申请应当自受理之日起60 日内决定是否同意,并通过药审中心网站通知申请人审批结果;逾期未通知的,视为同意。
药审中心已发布多项规范指南与技术指导原则,对上述程序和要求进行细化。针对变更是否可能影响受试者安全的具体评估标准,2020 年药审中心先后发布了《创新药(化学药)临床试验期间药学变更技术指导原则(征求意见稿)》和《临床试验期间生物制品药学研究和变更技术指导原则(征求意见稿)》,待其正式生效后即可成为申请人的评估依据。
对于试验申办者的评估与报告,药审中心在2020 年7 月1 日发布了《研发期间安全性更新报告管理规范(试行)》[12],规定申请人仅需按照要求准备、撰写和提交研发期间安全性更新报告,无需再提交《关于化学药IND 申请药学研究数据提交事宜的通知》中要求的化学药IND 申请药学研究年度报告。根据该规范,报告周期内发现的相关变更应分为3 类进行总结与报告:①药学的变化或者新发现,②非临床的变化或者新发现,③药物临床试验方案变更或者临床方面的新发现与报告,并对其中可能增加受试者安全性风险的变更进行补充申请。这为临床试验申请人在研发期间进行安全性更新报告提供了规范化依据。
相比之下,对于临床试验申办者主体变更的管理规定则不够清晰。《注册办法》第二十九条规定应该由变更后的申办者承担药物临床试验的相关责任和义务,但是并没有进一步细化规定对于不涉及上述药学、非临床或临床变更,仅是由于公司并购或产品转让而产生的申办者主体变更应该如何报告。建议尽快通过发布指南的方式,明确申办者主体变更的报告期限(立即、30 日或年报)、报告形式(由原申报者、变更后的申报者或共同报告),以及是否必须等待药审中心60 日的审评期限,还是可以在书面报告药审中心之后直接实施变更,而无需等待批准或反馈。
3.1.2 审评审批期间的变更
对于药品上市许可审评审批期间的变更,《注册办法》第四十条[2] 规定申请人对可能影响药品安全性、有效性和质量可控性的重大变更,应当撤回原注册申请,经补充研究后再重新申报。对于申请人名称变更、注册地址名称变更等不涉及技术审评内容的变更,申请人书面告知药审中心并提交相关证明材料即可。
对于由于公司并购或产品转让而导致的药品上市许可申请人主体变更,但又不涉及上述重大变更的情况,《注册办法》第四十条并没有进一步细化规定应该如何报告。建议尽快通过发布指南的方式,明确申请人主体变更的报告期限(立即、30 日或年报)、报告形式(由原申请人、变更后的申请人或共同报告),或者采用和上市后持有人主体变更相同的方式来管理。
3.2 药品上市后的变更管理
3.2.1 变更类别的确定和沟通
药审中心把药品上市后变更分为临床变更和药学变更。对于临床变更,经过2019 年11 月和2020 年4 月两次征求意见后,药审中心于2021年2 月发布《已上市化学药品和生物制品临床变更技术指导原则》[13],描述了如何对药品的适应症、适用人群范围、用法用量、药品说明书安全性信息、药物警戒计划等事项进行变更;如何根据临床变更的大小、对药品临床安全有效使用可能产生的影响及风险程度,将变更确定为重大、中等和微小变更;如何把重大变更细分为A 类和B 类,以及如何根据不同分类,执行对应的申报程序及技术要求。对于已上市药品增加境内未批准的新适应症或者改变给药途径,药审中心则规定必须按照药物临床试验和上市许可申请通道进行申报和审评审批。
对于药学变更,药审中心于2021 年2 月发布《已上市化学药品药学变更研究技术指导原则(试行)》[14],指出化学药品的药学变更包括制剂处方中辅料的变更、原料药和制剂生产工艺变更、生产场地变更、生产批量变更、制剂所用原料药的供应商变更、注册标准变更、包装材料和容器变更、有效期和贮藏条件变更,以及增加规格变更,列举了每种变更情形下的重大变更、中等变更、微小变更,以及需进行的研究验证工作,并以变更对药品的安全性、有效性或质量可控性产生影响的可能性作为标准进行分类。药审中心还于6月25 日发布《已上市生物制品药学变更研究技术指导原则(试行)》[15],列举了生物制品的原液、制剂以及按照生物制品管理的体外诊断试剂的常见变更事项,同样依据风险和产生影响的程度将各变更事项细分为重大变更、中等变更和微小变更,并明确了相应的技术要求。
国家药监局颁布的《变更办法》则规定了持有人根据上述技术指导原则进行研究验证之后,如果仍然无法确定变更类别,如何与药品监管部门进行沟通。境内持有人和境外持有人沟通程序有所不同。境内持有人可以与省级药品监管部门进行沟通,省级药品监管部门应当在20 日内书面答复,意见一致的按规定实施;对是否属于审批类变更意见不一致的,持有人应当按照审批类变更,向药审中心提出补充申请;对属于备案类变更和报告类变更意见不一致的,持有人应当按照备案类变更,向省级药品监管部门备案。境外持有人可以与药审中心沟通,具体沟通程序按照药品注册沟通交流的有关程序进行。
《变更办法》还规定了持有人可以根据管理和生产技术变化,与监管部门沟通是否可以降低技术指导原则中明确的变更管理类别,或降低持有人变更清单中的变更管理类别。境内持有人和境外持有人沟通降低管理类别的程序有所不同。境内持有人应当在充分研究、评估和必要验证的基础上与省级药品监管部门沟通,省级药品监管部门应当在20日内给予书面答复,意见一致的按规定执行,意见不一致的不得降低变更管理类别。境外持有人应当在充分研究、评估和必要的验证的基础上与药审中心沟通并达成一致后执行,意见不一致的不得降低变更管理类别。
3.2.2 持有人变更
根据《药品管理法》,持有人主体的变更属于审批类的重大变更。《变更办法》细分了三种持有人变更的情形:境内生产药品的持有人之间变更、境外持有人之间变更和已在境内上市的境外生产药品转移至境内生产的变更。其中,境内生产药品的持有人变更和境外持有人之间变更应由变更后的持有人向药审中心提出补充申请。对于已在境内上市的境外生产药品转移至境内生产的,应当由境内申请人按照药品上市注册申请的要求和程序提出新的药品注册申请,相关药学、非临床研究和临床研究资料如果适用时,可提交境外生产药品的原注册申报资料,符合要求的可申请成为参比制剂。
此外,持有人名称、生产企业名称、生产地址门牌号等不涉及主体事项的变更,《变更办法》规定应当在完成药品生产许可证相应事项变更后,向所在地省级药品监管部门就药品批准证明文件相应管理信息变更进行备案。境外生产药品上述信息的变更向药审中心提出备案。
值得注意的是,申请变更药品持有人的,药品的生产场地、处方、生产工艺、质量标准等应当与原药品一致;发生变更的,应该在持有人变更获得批准后,由变更后的持有人进行充分研究、评估和必要的验证,并按规定获得批准、进行备案或提交年度报告。
上述持有人变更管理制度与之前相比已经取得很大进步,但是仍有空间进一步细化完善,明确规定如何管理药品境内企业和境外企业相互转让药品上市许可的其他情形,包括如何进行境内生产境外持证,或者境外生产境内持证。
3.2.3 药品生产场地变更
《变更办法》规定变更药品生产场地的,药品的处方、生产工艺、质量标准等应当与原药品一致,持有人应当确保能够持续稳定生产出与原药品质量和疗效一致的产品。若是药品的处方、生产工艺、质量标准等发生变更,持有人应当进行充分研究、评估和必要的验证,并按规定获得批准、进行备案或提交年度报告。
境内持有人和境外持有人变更药品生产场地适用的程序有所不同。境内持有人或药品生产企业内部变更生产场地或境内持有人变更生产企业的,持有人(或药品生产企业)应当按照《生产办法》及相关变更技术指导原则要求进行研究、评估和必要的验证,向所在地省级药品监管部门提出变更《药品生产许可证》申请并提交相关资料。境外持有人变更药品生产场地且变更后生产场地仍在境外的,应按照相关技术指导原则进行研究、评估和必要的验证,向药审中心提出补充申请或进行备案。
3.2.4 其他生产变更
《变更办法》规定了其他药品生产变更,包括生产设备、原辅料及包材来源和种类、生产环节技术参数、质量标准等生产过程变更,以及原料药变更管理,持有人和原料药登记人应当充分评估该变更可能对药品安全性、有效性和质量可控性影响的风险程度,确定变更管理类别,按照有关技术指导原则和《药品生产质量管理规范》进行充分研究、评估和必要的验证,获得批准、进行备案或提交年度报告。在此规定的基础上,国家药监局与药审中心在2021 年2 月陆续发布了《已上市化学药品变更事项及申报资料要求》《化学药品变更受理审查指南(试行)》《已上市化学药品药学变更研究技术指导原则(试行)》及《已上市中药变更事项及申报资料要求》,进一步细化了化学药品与中药上市后变更的技术指导与申报资料要求。
《变更办法》还特别规定了在新修订《药品管理法》和《注册办法》实施前已发生的变更的处理方式,指明在以下两种情况下,无需按照新的变更管理规定及技术要求进行重新申报:①持有人或生产企业按照原生产工艺变更管理的有关规定和技术要求经研究和验证证明不影响药品质量的已实施的变更;或②经过批准或再注册中已确认的工艺。上述规定为企业处理《药品管理法》和《注册办法》实施前已发生的变更提供了依据,避免重复申报。
3.2.5 变更执行程序和监管
对于属于审批类的重大变更和报告类的微小变更,《变更办法》的规定比较明确:审批类变更应当由持有人向药审中心提出补充申请,按照有关规定和变更技术指导原则提交研究资料,经批准后实施,审评工作时限按照《注册办法》有关规定执行;报告类变更则应当由持有人按照变更管理的有关要求进行管理,在年度报告中载明。
对于备案类变更,《变更办法》规定应当由持有人向药审中心或省级药品监管部门备案,备案部门应当自备案完成之日起5 日内公示有关信息,省级药品监管部门应当根据备案变更事项的风险特点和安全信用情况,自备案完成之日起30 日内完成对备案资料的审查,必要时可实施检查与检验。对于持有人何时必须提交备案,以及何时视为完成备案可以实施变更,上述规定不够明确,但是同时颁布的《变更办法》的政策解读则指出备案不是行政许可,持有人按照备案资料要求提交备案资料后即完成备案,可以立即实施相应变更。
可以立即实施的解读似乎有悖于上述省级药品监管部门在30 日内对备案资料进行审查的规定,建议此处细分为两种备案时限:①备案完成后即可立即实施变更的备案,以及②备案完成后必须等待,给予省级药品监管部门30 日完成上述对备案资料的审查之后方可实施变更的备案。
《变更办法》规定药品监管部门在监管过程中,发现持有人已实施的备案或报告类变更的研究和验证结果不足以证明该变更科学、合理、风险可控,或者变更管理类别分类不当的,应当要求持有人改正并按照改正后的管理类别重新提出申请,同时对已生产上市的药品开展风险评估,采取相应风险控制措施。
04、违反药品变更管理规定的法律责任
临床试验和审评审批期间如果违反药品变更管理规定,申请人必须承担相应法律责任。如果临床试验期间未按照相关规定进行补充申请,可能构成《药品管理法》(2019)第一百二十五条[1] 规定的“未经批准开展药物临床试验”,法律责任包括暂停临床试验,没收违法所得以及包装材料、容器,责令停产停业整顿,罚款和准入限制等。此外,在临床试验期间或药品注册过程中,如果故意隐瞒变更,可能被判定为提供虚假的证明、数据、资料、样品或者采取其他手段骗取临床试验许可或者药品注册等许可,面临撤销相关许可、十年内不受理其相应申请、罚款和拘留等法律责任。
《药品管理法》(2019)第一百二十四条和第一百二十七条[1] 分别规定了未经批准在药品生产过程中进行重大变更和未按照规定对药品生产过程中的变更进行备案或者报告的法律责任:①未经批准在药品生产过程中进行重大变更的法律责任包括没收药品和违法所得以及专门用于违法生产的原料、辅料、包装材料和生产设备,责令停产停业整顿,并处违法药品货值金额十五倍以上三十倍以下的罚款;货值金额不足十万元的,按十万元计算;情节严重的,吊销药品批准证明文件直至吊销药品生产许可证、药品经营许可证或者医疗机构制剂许可证,对法定代表人、主要负责人、直接负责的主管人员和其他责任人员,没收违法行为发生期间自本单位所获收入,并处所获收入百分之三十以上三倍以下的罚款,十年直至终身禁止从事药品生产经营活动,并可以由公安机关处五日以上十五日以下的拘留。②未按照规定对药品生产过程中的变更进行备案或者报告的法律责任包括责令限期改正,给予警告;逾期不改正的,处十万元以上五十万元以下的罚款。此外,《生产办法》第六十八条和第七十一条[4] 还规定了生产企业未经批准变更生产地址、生产范围的责任,以及变更企业名称、住所、法定代表人未按规定办理登记事项变更的责任,包括责令关闭、没收违法所得、罚款等。
药品上市后如果违反变更管理规定,还可能构成假药、劣药。根据《药品管理法》(2019)第九十八条[1] 的规定,假药包括药品所含成份与国家药品标准规定的成份不符等药品;劣药则指药品成份的含量不符合国家药品标准的规定等。因此,如果药品变更导致药品的成份或含量不符合国家药品标准,可能造成药品被认定为假药或劣药。《药品管理法》(2019)第一百一十六条至第一百一十八条[1] 规定了生产、销售假药、劣药的法律责任,包括对企业和相关责任人员没收违法所得,责令停产停业整顿,吊销许可证,罚款,准入限制等处罚。
05、结 语
我国在2019 年《药品管理法》与2020 年《注册办法》的框架下,规定了药品变更的分类管理原则,确定了审批、备案和报告的事项范围,以及相应的提交和沟通程序,取得了很大进步。对于各种变更的具体评估标准与技术要求,国家也出台了相应的指南文件,使得我国药品变更管理更加体系化、科学化,着重考量变更对于药品安全性、有效性和质量可控性的可能影响,依据风险的不同,规定了适当的管控,形成了企业和政府有法可依、管理有效的局面。
当然,我国的药品变更管理法律体系尚可进一步发展和完善,一些问题有待政策的落地和配套措施的出台,例如对于药品上市后变更,虽然《变更办法》已经有了很多详细规定,但是国家统一的变更申报系统和各省的备案细则尚未出台,境内企业和境外企业相互转让药品上市许可的程序尚未明确,境内持有人与省级药品监管部门就确定变更类别、降低变更类别如何沟通的具体程序也有待各省级药品监管部门制定。对于药品上市前变更,则缺乏类似《变更办法》的详细规定。期待药品变更管理法律体系会伴随我国药品监管的发展与改革,进一步得以完善。
·引用本文
陈少羽,王文佳*,戴安迪,杨语涵.药品变更管理法律体系的发展与改革[J].中国食品药品监管.2021.07(210):84-91.
药品变更管理法律体系的发展与改革
Development and Reform of the Legal System for Drug Change Management in China
陈少羽
美国安诺波特律师事务所上海办公室
CHEN Shao-yu
Arnold & Porter Shanghai Representative Office
王文佳*
美国安诺波特律师事务所上海办公室
WANG Wen-jia*
Arnold & Porter Shanghai Representative Office
戴安迪
美国安诺波特律师事务所上海办公室
DAI An-di
Arnold & Porter Shanghai Representative Office
杨语涵
美国安诺波特律师事务所上海办公室
YANG Yu-han
Arnold & Porter Shanghai Representative Office
·作者简介
陈少羽,美国安诺波特律师事务所上海办公室管理合伙人。专业方向:生命科学与医疗监管
王文佳,美国安诺波特律师事务所上海办公室资深顾问。专业方向:生命科学与医疗监管、并购、外商直接投资、劳动法、公司治理和合规

来源:中国食品药品监管杂志


