您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-10-31 21:11
新修订《医疗器械监督管理条例》(以下简称新条例)对医疗器械临床评价相关要求进行了全面修订,其中关于临床评价及临床试验的相关变化主要有:规定临床试验审批实行默示许可;厘清临床评价和临床试验的关系,明确临床评价管理要求;鼓励临床机构开展创新医疗器械临床试验;增加拓展性临床试验规定等。
随着新条例的正式施行,与医疗器械临床评价相关的配套规章陆续于近期发布,如《医疗器械注册与备案管理办法》和《体外诊断试剂注册与备案管理办法》,《医疗器械临床评价技术指导原则》《医疗器械临床评价等同性论证技术指导原则》《决策是否开展医疗器械临床试验技术指导原则》《医疗器械注册申报临床评价报告技术指导原则》《列入免于进行临床评价医疗器械目录产品对比说明技术指导原则》等。
医疗器械临床评价法规实施进展
9月以来,国家药监局陆续发布了与医疗器械临床评价相关的法规,如《免于临床评价医疗器械目录》《免于临床试验体外诊断试剂目录》,《免于临床试验的体外诊断试剂临床评价技术指导原则》《体外诊断试剂临床试验技术指导原则》《医疗器械临床评价技术指导原则》《医疗器械临床评价等同性论证技术指导原则》《决策是否开展医疗器械临床试验技术指导原则》《医疗器械注册申报临床评价报告技术指导原则》《列入免于进行临床评价医疗器械目录产品对比说明技术指导原则》等。其中,还在征求意见中的有《医疗器械临床试验数据递交要求注册审查指导原则》《医疗器械临床试验质量管理规范》《医疗器械临床试验方案范本》等。
除此之外,在新条例发布前已经开始实施的与临床评价相关的法规包括《医疗器械拓展性临床试验管理规定(试行)》《需进行临床试验审批的第三类医疗器械目录(2020年修订版)》《接受药品境外临床试验数据的技术指导原则》《医疗器械临床试验检查要点及判定原则》《国家食品药品监督管理总局关于开展医疗器械临床试验监督抽查工作的通告》、《国家食品药品监督管理总局关于医疗器械临床试验备案有关事宜的公告)》等。
医疗器械临床评价政策演变
在此轮医疗器械法规修订中,国家药监部门根据IMDRF临床评价系列协调文件,将医疗器械临床评价的定义与国际接轨。将原有的免于临床试验的评价方式规范为免于临床评价,为非临床试验的临床评价提供更多更细的评价方法和原则,对需要进行临床试验的情况进行了更细致的分析。
1.免于进行临床评价
从定义规范来看,本次修订的法规认为仅做等同性论证的行为不算临床评价,那么原先我们认为的列入免于进行临床试验的医疗器械目录,则更新为免于进行临床评价的医疗器械目录。从目录内容上来看,本次发布的《免于进行临床评价的医疗器械目录》是2018年12月20日发布的修订版《免于进行临床试验的医疗器械目录》和2021年1月发布的《免于进行临床试验医疗器械目录(第二批修订)》的汇总版本,条款内容没有发生改变,范围没有发生变化。
2.需要进行临床评价
《医疗器械注册申报临床评价报告技术指导原则》指出,对于未列在《免于进行临床评价的医疗器械目录》中的产品,需要进行临床评价,而临床评价的手段可以是通过开展临床试验,或者通过对同品种医疗器械临床文献资料、临床数据进行分析评价。其中,对同品种医疗器械临床数据进行分析、评价时可以采用“等同器械的临床数据进行临床评价”或者“使用可比器械的临床数据进行部分临床评价”。对临床试验数据进行分析、评价时,“临床试验包括在中国境内开展的临床试验,在中国境外开展的临床试验、多区域临床试验以及临床文献报道的临床试验”。“注册人可根据申报产品的技术特征、适用范围、已有临床数据等具体情况,选择恰当的评价途径或者评价路径的组合,开展临床评价”。
3.体外诊断试剂临床评价
自2017年10月30日发布第一批《免于进行临床试验的体外诊断试剂目录》和2017年11月3日出台了《免于进行临床试验的体外诊断试剂临床评价资料基本要求(试行)》,体外诊断试剂开启了免于进行临床试验的通道。2021年9月24日,《免于临床试验的体外诊断试剂临床评价技术指导原则》已发布。
对于传统的需要进行临床试验的体外诊断试剂,也已经发布了新版《体外诊断试剂临床试验指导原则》。很明显的不同是,新版《体外诊断试剂临床试验指导原则》不再规定体外诊断试剂临床试验的样本量,而是采用统计学要求进行计算,要求注册人“根据产品特点和产品性能评价需要,体外诊断试剂临床试验可能包括不同的临床试验目的,有必要针对各个临床试验目的,分别进行科学的临床试验设计,包括选择适当的临床试验设计类型,确定适合的对比方法、受试者入组/排除标准和临床评价指标等,并进行科学的样本量估算。”
临床评价法规变化对行业的影响
根据新发布的《医疗器械临床评价指导原则》《医疗器械注册申报临床评价报告技术指导原则》《医疗器械临床评价等同性论证技术指导原则》,医疗器械的临床评价工作将扩展到“医疗器械全生命周期”。
对于《列入免于进行临床评价医疗器械目录产品对比说明技术指导原则》要求提交的相应资料,从指导原则内容上来看,与2015年发布的《医疗器械临床评价技术指导原则》中列入《免于进行临床试验的医疗器械目录》产品的临床评价要求基本一致。
对于需要进行临床评价的医疗器械产品,需从企业自身情况出发,根据产品特征、临床风险、已有临床数据等情形,充分考虑资源的掌握情况,选择合理的临床评价路径。一般有以下几种情况:
1)通过同品种医疗器械临床文献资料、临床数据进行评价可以证明产品的安全有效性,可以采用单纯用同品种对比的形式进行临床评价;
2)申报产品或申报产品的前代产品,已有部分临床文献,临床数据,该数据包括中国境内开展的临床试验,在中国境外开展的临床试验、多区域临床试验以及临床文献报道的临床试验,或者已有同类器械的临床数据均不足以确认产品安全有效的情况下,应开展对无法确认的部分的临床试验。若已有临床数据为申报产品的境外临床数据、临床经验数据等,可开展基于原有临床试验方案的补充临床试验。若已有临床数据为同品种产品的数据,则可开展针对差异性的临床试验。
3)若企业无法掌握任何申报产品或申报产品前代产品的临床资料,也无法掌握同类产品的临床资料,则应当开展临床试验。
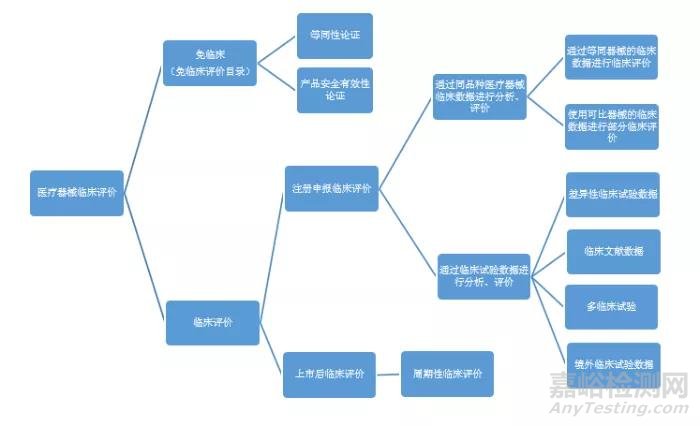
图:医疗器械临床评价路径
总体而言,新条例及其配套法规重新调整了医疗器械临床评价的概念,为申请人提供了更多的临床评价思路。从注册人角度来看,新法规鼓励注册人在产品研发时更加关注行业发展和行业技术水平,在临床使用方面做好充分调研,以掌握更多的产品、技术、资源。注册人只有掌握大量的数据和资源,才能对临床评价作出更科学的决策。

来源:中国体外诊断网 CAIVD


