您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-07-25 12:19
药物共晶是改善难溶性药物溶出与生物利用度有效的策略之一, 但其在溶出过程中容易发生相转变导致原料药重结晶, 失去增溶优势, 该问题已成为难溶性药物共晶应用的主要瓶颈之一。选择合适的添加剂能够有效抑制药物共晶溶出过程中的相转变, 提高难溶性药物共晶溶出与吸收程度; 此外, 复杂的胃肠道给药环境与药物共晶体内吸收行为, 均会对药物共晶的生物利用度产生明显影响。因此, 本文总结了关于难溶性药物共晶溶出与吸收的研究进展, 期望为药物共晶制剂处方的开发提供指导。
关键词
药物共晶; 难溶性药物; 相转变; 体外溶出; 体内吸收
目前, 大约有40%的上市药物与90%的候选化合物属于难溶性分子, 这一现状影响了药物的开发及临床应用, 因此, 解决药物难溶性问题成为药物研究的重点与挑战之一。据统计, 已上市的药品中有80%以上的药物以固体制剂的形式存在, 包括散剂、颗粒剂、滴丸剂、胶囊剂和片剂等。在制剂开发过程中, 研究者通过改变固体制剂中的药物固态形式, 从而影响药物的溶解度、吸湿性、稳定性、生物利用度、粉体学性质(如可压性、流动性)等, 例如将药物制备成无定形固体分散体、溶剂合物、盐型、药物共晶等固态形式。无定形固体分散体(amorphous solid dispersion, ASD)可使药物无定形化后处于高能态, 同时溶出过程中无定形药物无需克服晶态药物溶出时所需的晶格能, 因此有利于药物溶出, 但无定形药物可能在储存或溶出过程中发生表面结晶或表面相分离等现象, 降低其物理稳定性, 进而不利于药物溶出。药物制备成盐型是解决离子型药物低溶解度与低生物利用度有效的手段之一, 但能够成盐的药物仅适用于离子型药物, 故药物成盐的使用对象有一定局限。
药物共晶是指在同一晶格中包含两种或两种以上不同分子(其中一种是活性药物成分)以固定的化学计量比通过非离子键和非共价键结合形成的晶体, 如图1A所示, 形成共晶的非共价键主要包括氢键、π-π堆积作用、范德华力等弱相互作用力, 由于氢键本身键能较大且具有方向性, 故在共晶形成中占据主要地位。
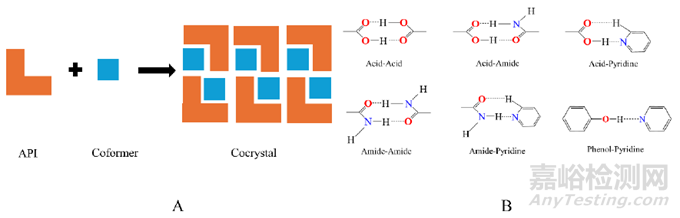
Figure 1 Cocrystal structure introduction. A: A pharmaceutical cocrystal is a stoichiometric molecular complex of an API molecule (orange) with a coformer (blue) assembled via noncovalent interactions, predominantly hydrogen bonds; B: Common supramolecular synthons in crystal engineering. API: Active pharmaceutical ingredient
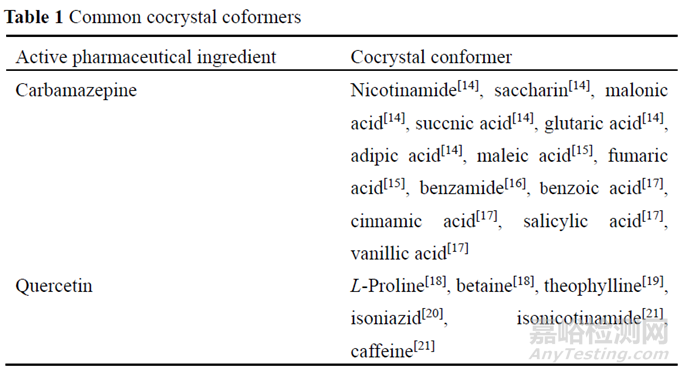
美国食品和药物管理局(Food and Drug Administration, FDA)与欧洲食品和药物管理局(European Medicines Agency, EMA)对共晶的定义有所不同, FDA将共晶分类为“溶剂化物的一种特殊情况, 且第二组分(配体)不是溶剂”, 并认为共晶不属于新的活性物质; 而EMA指出“共晶与盐具有许多概念上的相似之处, 溶剂合物应视为共晶的其中一类”, 并且EMA认为如果某种共晶对药物的疗效与安全性有所改善, 应视为新的活性物质。共晶配体的选择通常为安全无毒性物质, “一般公认安全”(generally recognized assafe, GRAS)清单中的化合物均可作为药学上可接受的共晶配体, 常用的共晶配体如表1所示(以卡马西平共晶和槲皮素共晶为例)。
药物共晶通过向原料药分子中引入共晶配体分子, 从而改变原料药分子的排列与堆积方式, 并改变原料药的相互作用模式; 从晶体工程设计的角度出发看, 药物共晶的形成包含两个过程: 首先, 药物与配体分子通过氢键或其他相互作用识别形成超分子合成子; 其次, 超分子合成子堆积组装形成药物共晶。超分子合成子指结构单元通过相互作用而组装成的超分子, 超分子合成子在共晶合成中发挥着重要作用, 共晶中常见的超分子合成子有羧基−羧基、羧基−酰胺、羧酸−芳香氮、酰胺−酰胺、酰胺−芳香氮和酚羟基−芳香氮等(图1B)。
药物共晶的形成可以通过热分析、光谱学与波谱学等多种手段分析表征鉴定。差示扫描量热分析法(differential scanning calorimetry, DSC)可以用于测定固态物质的熔点, 当药物与配体合成共晶后, 其熔点通常与药物和配体的单组分熔点有明显区别, 但仅使用DSC无法确保共晶合成, 因为药物与配体可能形成低共融混合物, 或在DSC加热过程发生共结晶, 因此该方法需要联合其他表征手段用以确定共晶的合成。拉曼光谱(raman spectroscopy, Raman)通过检测入射光的散射程度可以分辨出共晶与药物分子振动变化的差异, 共晶的谱峰相较药物的谱峰会产生位移、形状变化或产生新的谱峰等现象, 且该技术具有对样品无损、需样量少、快捷方便等优势。X-射线粉末衍射(powder X-ray diffraction, PXRD)图谱可以看作不同分子晶体结构的“指纹图谱”, 共晶的PXRD图谱上的特征峰会与药物和配体的特征峰产生明显区别。药物共晶与盐的鉴别方法主要通过单晶X射线衍射技术(single crystal X-ray diffraction, SCXRD), 该技术可获得晶体结构包括键长、键角、分子堆积结构等全部三维信息, 能够判断组分之间是否存在质子转移或形成氢键, 通过单晶X射线衍射技术可以得到盐和共晶羧酸基团的ΔDC−O(C−O与C=O键长的距离差), 若ΔDC−O<0.03Å, 则该组合为盐, 若ΔDC−O>0.08Å, 则为共晶。
1、药物共晶的优势
药物共晶在不改变药物分子化学结构的基础之上, 能够调控难溶性药物的溶解度与溶出速率。例如芹菜素−茶碱共晶与大豆黄酮−茶碱共晶相较原料药的固有溶出速率分别提升约8.4与2.3倍, 帕博西尼−间苯二酚共晶在pH6.8下溶出24h后的浓度约为帕博西尼原料药的3倍。药物共晶还能改善药物的稳定性、机械性质、生物学性质等, 例如McKellar等通过合成丙泊酚−异烟酰胺共晶, 显著提高丙泊酚熔点, 使其在常温下由液态形式转为固态形式, 大大提升药物的热稳定性; Wang等研究发现替莫唑胺−橙皮素共晶改善了替莫唑胺的塑性与可压性, 原因在于替莫唑胺晶体呈不利于压片的网状结构, 而替莫唑胺−橙皮素共晶晶体结构呈层状从而增强了可压性; 奥沙利铂−山奈酚共晶相较奥沙利铂而言, 在40℃/75%RH的加速稳定性实验条件下, 表现出更好的物理稳定性, 同时该共晶对乳腺癌细胞活性有明显抑制作用, 可能由于该共晶能够有效调控奥沙利铂溶出行为并且奥沙利铂和山奈酚联用会实现增效。
2、药物共晶溶出模型与相转变
为了提高难溶性药物的溶出, 一般会选择水溶性高的共晶配体与之形成共晶。由于药物与配体之间的氢键相互作用较弱, 在溶出过程中, 亲水性配体更容易先从共晶晶格中逃逸并快速在水溶液中溶解, 同时促进难溶性药物分子的溶出, 上述过程即为药物共晶溶出时的“弹簧”效应; “弹簧”效应下形成的药物过饱和溶液属于热力学不稳定体系, 此时过饱和的药物分子会倾向在溶液中重新结晶析出, 由共晶形式转为难溶性原料药晶体析出的过程称为相转变, 从而导致药物共晶失去增溶优势, 这一过程即为药物共晶溶出时的“降落伞”效应, 以上溶出过程统称为“弹簧—降落伞”模型, 如图2所示。如He等发现山奈酚-L-脯氨酸共晶溶出会在15min以内达到近4倍的山奈酚平衡溶解度, 并在60min以内下降至约2倍的平衡溶解度, 6h内稳定维持该浓度。
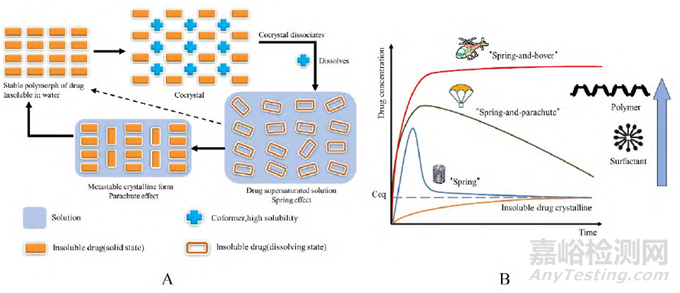
Figure 2 Cocrystal dissolution mechanisms and models. A: Cocrystal dissolution mechanisms; B: Drug concentration-time profile during cocrystal dissolution
这种共晶溶出模型也并非适用于所有药物共晶, 例如Stanton等考察AMG517与多种不同配体的共晶溶出行为仅存在“弹簧”过程, 药物浓度达到峰浓度以后快速下降, 可能是由于药物从溶液中快速析晶所致, 这一过程统称为“弹簧”模型(图2A)。除此之外, 药物共晶溶出后药物浓度也可能会达到高浓度平台并长时间维持, 该过程被称为“弹簧——直升机”模型, 该模型可用于解释溶出后可形成药物−配体复合物形式的共晶体系, 例如布洛芬−烟酰胺共晶在溶出时布洛芬能够达到高浓度的过饱和溶液状态并能够长时间维持稳态, 其原因在于布洛芬与烟酰胺在溶出过程中形成了溶解度更高的水溶性复合物, 可确保药物浓度长时达到高浓度平台。另一方面, 有文献报道通过计算模拟手段证明农药二氯苯氧乙酸−吩嗪共晶溶解以后药物会与配体在溶液中通过氢键作用形成分子簇, 这可能也是该共晶能够维持药物溶出过饱和现象的原因所在。
为了抑制共晶溶出中的相转变, 在溶出介质中加入添加剂可较好维持药物共晶的过饱和状态, 使溶液体系尽可能达到稳态, 并使原本在溶出过程中处于“弹簧”或“弹簧—降落伞”的共晶溶出趋势有可能转变为“弹簧—直升机”的溶出趋势, 从而更好发挥共晶增溶优势(图2B)。
上述在共晶溶出过程中由共晶形式又转为难溶性原料药晶体析出的过程称为溶液介导的相转变(solution-mediated phase transformation), 溶液介导相转变主要包括两种途径, 包括体相溶液介导的相转变(bulk phase solution-mediated phase transformation, BP-SMPT)和颗粒表面溶液介导的相转变(particle surface solution-mediated phase transformation, PS-SMPT), 药物共晶溶出时高水溶性的共晶配体接触水溶液从而脱离共晶晶格束缚, 此时难溶性药物分子会在局部形成高过饱和并诱导难溶性药物析晶, 在共晶表面形成局部高过饱和态并发生析晶现象为PS-SMPT, 在体相溶液中形成局部高过饱和并发生析晶现象为BP-SMPT。Omori等研究了卡马西平−戊二酸(carbamazepine-glutaric acid, CBZ-GLA)共晶溶出过程中的相转变行为, 该共晶溶出曲线显示, 其与卡马西平二水合物(CBZ dihydrate, CBZDH)溶出浓度相似, 通过PXRD与DSC研究显示溶出10min后CBZ-GLA共晶基本完全转为CBZDH, 溶出实验与固相表征共同证明CBZ-GLA共晶发生了表面相转变行为, 偏光显微镜与扫描电镜对溶出残余物的观察进一步佐证CBZ-GLA共晶表面相转变行为的发生。
综上所述, 药物共晶具有明显改善溶出等诸多优势, 但溶出过程中又存在较为复杂的相转变问题, 且药物共晶配体的选择、聚合物与表面活性剂的选择均对体外溶出会有较大的影响, 而药物的渗透方式、胃肠道pH与生物活性物质的存在都会影响共晶的溶出与吸收行为。因此, 研究药物共晶溶出与吸收的影响因素对药物共晶制剂的开发具有重要意义。
3、难溶性药物共晶溶出的影响因素
3.1 共晶自身性质对药物共晶溶出的影响
共晶自身性质如粒径、表面性质等因素会对药物共晶溶出产生影响。Pi等成功制备黄芩素−烟酰胺纳米共晶, 在模拟肠液环境(fasted state simulated intestinal fluid, FaSSIF)中, 该纳米共晶在30min内快速溶出后基本达到溶出平台期, 并在360min内能够维持浓度稳定, 而黄芩素−烟酰胺共晶在360min内持续缓慢溶出, 并且最终溶出浓度低于纳米共晶, 说明共晶的粒径会显著影响共晶的溶出行为; 共晶的表面性质也是影响共晶溶出差异的原因之一, 例如根皮素−烟酰胺共晶和根皮素−异烟酰胺共晶中根皮素分子表面的π-π作用力与氢键作用力占比有所不同, 这可能是影响共晶溶出行为差异的原因之一; 酮康唑−富马酸共晶在溶出之后的界面pH会显著低于酮康唑溶出的界面pH, 溶出界面pH的降低可能是促进酮康唑组分从共晶中溶出的原因之一。
由于晶体的不同晶面存在特异性, 且晶体表面的粗糙度会影响其表面的润湿性, 可能会进一步导致不同晶面上的共晶相转变行为也会存在显著差异。Omori等研究了卡马西平−糖精(carbamazepine-saccharin, CBZ-SAC)共晶单晶表面相转变行为, 发现在与缓冲液接触后, CBZDH在CBZ-SAC共晶(010)晶面表面上快速结晶, 但在(001)晶面上几乎不结晶, 这可能与(010)晶面的粗糙度(7.165μm)大于(001)晶面的粗糙度(1.475μm)有关。
3.2 配体对药物共晶溶出的影响
3.2.1 不同配体对药物共晶溶出的影响
同一个药物可以与多种配体形成共晶, 在相同的溶出条件下, 配体不同的共晶通常呈现出不同的溶出行为。如黄芩素可以与异烟酰胺、异烟肼、茶碱与咖啡因等多种配体形成共晶, 但黄芩素−咖啡因共晶改善溶出优势明显优于黄芩素−茶碱共晶, 黄芩素−茶碱共晶对黄芩素溶出几乎无明显改善作用; Guo等探究氟芬那酸−烟酰胺(flufenamic acidnicotinamide, FFA-NIC)共晶与氟芬那酸−茶碱(flufenamic acid-theophylline, FFA-TP)共晶溶出机制不同, FFA-NIC共晶溶出时会发生共晶表面相转变, FFATP共晶溶出时会发生体相相转变; Omori等发现CBZ分别与顺反异构体马来酸(maleic acid, MAL)和富马酸(fumaric acid, FUM)合成的共晶在相同溶出条件下, CBZ-FUM共晶没有发生表面相转变且溶出速率较快, CBZ-MAL共晶发生了表面相转变且溶出速率较慢; 抗病毒药阿德福韦酯(adefovir dipivoxil, ADD)与烟酰胺(nicotinamide, NIC)和糖精(saccharin, SAC)分别形成的共晶在水中的溶解度差异较大, ADD-SAC共晶溶解度约为ADD溶解度的4倍, ADD-NIC共晶未明显改善ADD溶解度, 原因在于弱酸性配体SAC溶出后使溶液pH显著降低, 从而促进弱碱性药物ADD的溶解。可见配体的选择对药物共晶的溶出行为有十分重要的影响。
对于难溶性药物, 通常应选择水溶性配体来提高药物溶解度和溶出速率, 例如黄酮类化合物根皮素分别和水溶性优良的NIC与异烟酰胺形成不同共晶, 均明显改善了根皮素的溶解度和溶出速率; 但有研究发现共晶溶出过程中溶液的稳定性与配体溶解度不呈正相关关系, Yoshimura等发现抗血小板聚集药西洛他唑与3种苯甲酸衍生物配体形成的共晶, 配体的溶解度越高, 共晶在溶出过程中相转变速率越快, 导致共晶的溶出优势越小。
共晶自身的晶体结构也是影响其溶出行为的一大因素, 例如降血糖药依帕司特与甜菜碱形成共晶后会形成药物与配体交错排列的层状堆积结构,利于依帕司特的溶出, 共晶中药物的溶出速率达原料药的3.5倍左右; CBZ-NIC共晶对溶出的改善能力与抑制相转变能力均优于CBZ-GLA共晶, 原因在于CBZ-GLA 共晶的分子排布与CBZDH晶体的分子排布更为相似, 容易发生相转变, 从而促进共晶相转变的趋势, 导致溶出速率较低。
3.2.2 过量配体对药物共晶溶出的影响
在溶出介质中加入过量配体会影响药物共晶的体外溶出速率与维持过饱和行为, 针对CBZ-GLA共晶的固有溶出速率测定实验而言, 采用不同比例的共晶与配体混合物, 药物溶出速率大小顺序为: CBZ-GLA/GLA(1∶3)>CBZ-GLA/GLA(1∶1)>CBZ-GLA/GLA(3∶1), 上述结论显示, 加入过量配体会提升共晶中药物的固有溶出速率。Kataoka等研究发现针对抗菌药酮康唑-4-氨基苯甲酸共晶而言, 配体在非漏槽粉末溶出下溶液中的预溶量从0.474mmol·L-1逐渐增至15.2mmol·L-1的过程中, 共晶溶出行为发生明显变化, 同时当配体投入量大于7.58mmol·L-1时, 药物维持的过饱和浓度逐渐降低, 原因在于过量配体会使药物溶出的瞬时浓度低于临界成核浓度以避免“降落伞”过程, 从而能够长时间维持共晶溶出的过饱和状态, 但配体浓度过高时, 由于共晶溶解度的限制, 药物溶出能达到的最大浓度也会随之降低(图3)。
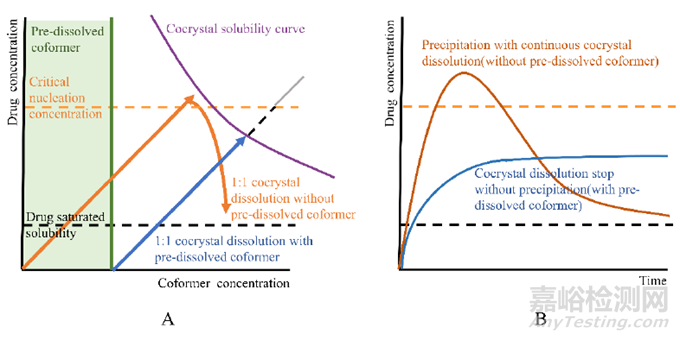
Figure 3 Theoretical cocrystal dissolution mechanisms with /without pre-dissolved conformer. A: Drug concentration-coformer concentration profile; B: Drug concentration-time profile
3.3 高分子聚合物对药物共晶溶出的影响
高分子聚合物是药物共晶溶出中常用的相转变抑制剂, 例如难溶性药物伊曲康唑与琥珀酸形成的共晶在溶出5min后发生明显的相转变, 维持溶出过饱和能力非常弱, 而当在溶出介质中加入一定量的纤维素类高分子或聚维酮类高分子抑制共晶在溶液中的相转变, 可使药物维持过饱和能力显著提升; Shigemura等发现添加剂对共晶体相相转变与表面相转变的抑制作用并不相同, 其主要原因可能在于具有较多疏水基团的辅料羟丙甲基纤维素(hydroxypropyl methylcellulose, HPMC)、醋酸羟丙甲纤维素琥珀酸酯(hydroxypropylmethyl cellulose acetate succinate, HPMCAS)与氨基甲基丙烯酸酯共聚物(amino methacrylate copolymer, Eudragit EPO)更容易吸附在疏水性药物CBZ晶体表面而抑制晶体生长, 而相对亲水的辅料聚乙二醇(polyethylene glycol, PEG)、聚乙烯基吡咯烷酮(polyvinylpyrrolidone, PVP)与甲基丙烯酸共聚物(methacrylic acid copolymers, Eudragit L100-55)对CBZ晶体的吸附作用不强, 没有良好的抑制晶体生长能力。而Jasani等研究发现羟丙纤维素(hydroxypropylcellulose, HPC)不能抑制依西美坦−马来酸(Exemestane-maleic acid, EXE-MAL)共晶的相转变, 但是却能维持共晶溶出的过饱和态, 推测可能是HPC通过与共晶溶出相转变后产生的依西美坦晶粒发生相互作用后吸附在晶粒表面, 抑制了依西美坦晶粒的生长而使纳米晶粒二次再溶出(图4)。同时在药物成核生长的相关研究中也发现, 高分子还可以通过增加溶液粘度和增加空间位阻等方式来限制药物分子的运动从而抑制药物成核。
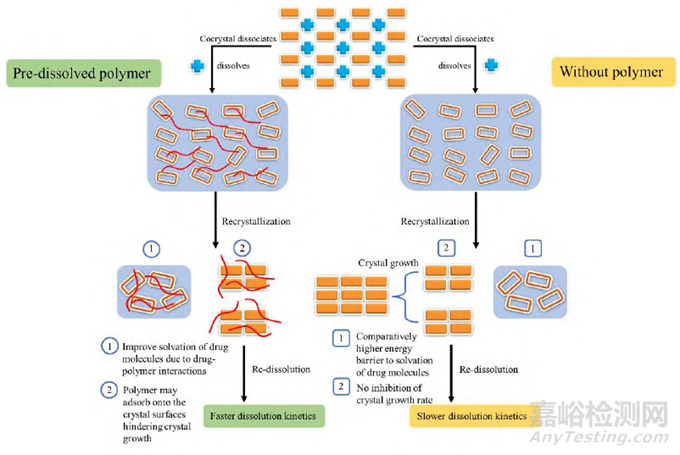
Figure 4 EXE-MAL cocrystal dissolution mechanism profile. EXE-MAL: Exemestane-maleic acid
许多研究已经证实, 药物共晶溶出过程中, 高分子聚合物对共晶溶出时难溶性药物相转变的抑制和溶出过程中过饱和度的维持通常是通过药物与聚合物的相互作用来实现的。高分子聚合物主要通过氢键与共晶组分发生相互作用, Guo等在研究添加剂PVP与乙烯基吡咯烷酮/乙酸乙烯酯共聚物(polyvinylpyrrolidone vinylacetate copolymer, PVPVA)对FFA-NIC共晶和FFATHP共晶的溶出和渗透的影响的实验中, 通过一维核磁共振氢谱证明聚合物PVP和PVPVA与药物和配体存在分子间相互作用, 从而影响共晶的溶出与渗透行为; Zhang等通过二维核磁探究发现沙库巴曲−缬沙坦共晶与PVP、HPMC、HPC等存在疏水相互作用, 并通过相互作用影响共晶溶出与渗透行为;Kirubakaran等通过原子力显微镜观察、扫描电镜观察与分子动力学模拟等手段探究PVP、PVPVA与PEG对FFA-TP、FFA-NIC与CBZ-NIC共晶的相互作用, 证明3种聚合物对3种共晶表面相转变的抑制效果均为: PVP>PVPVA>PEG, PVP抑制共晶相转变效果更好的原因可能在于PVP在共晶表面形成弯曲的吸附构象并形成较高的静电相互作用, 降低了PVP在共晶表面的运动性, 使得PVP在共晶表面易形成位阻层, 从而更好抑制共晶相转变。
高分子聚合物在ASD中作为基质也会通过抑制相转变行为等途径影响药物溶出与吸收行为, 与对于药物共晶的作用机制基本相似, 但高分子聚合物在无定形固体分散体制备过程中和无定形药物已经产生较强的相互作用, 从而也导致ASD的载药量不同会有不同的溶出机制, 另外高分子聚合物对药物共晶的作用主要体现在共晶溶出以后, 而对ASD而言, 高分子聚合物不仅会抑制ASD溶出以后的相转变行为, 同时会在ASD的固态形式中抑制无定形药物的相转变行为, 这与药物共晶有所不同。
3.4 表面活性剂对药物共晶溶出的影响
表面活性剂可以通过增加药物的平衡溶解度, 从而降低药物共晶溶出后溶液的过饱和度, 使共晶溶解优势即共晶溶解度与药物溶解度的比值SA(solubility advantage, SA=SCC/SD, SCC指共晶的平衡溶解度, SD指药物的平衡溶解度)值降低。SA值越低, 药物共晶与药物本身的溶解行为差异越小, 会从热力学上抑制共晶相转变, 使共晶溶出能够更好维持过饱和行为。例如, Huang等发现聚氧乙烯20油醚[polyoxyethylene(20) oleyl ether, Brij99]、十二烷基硫酸钠(sodium lauryl sulfate, SLS)和聚氧乙烯40硬脂酸酯[polyoxyethylene(40) stearate, Myrj52]可以不同程度促进吲哚美辛−糖精(indomethacin-saccharin, IMC-SAC)共晶溶出, 当SA值越低时, 表面活性剂越能够促进IMC-SAC共晶溶出; 当投入7.78mmol·L-1Brij99时SA=2, 此时共晶溶出维持过饱和能力最强, 3h内没有发生共晶相转变。
另外, Cavanagh等以达那唑−香草酸为模型共晶, 通过在表面活性剂聚乙二醇1000维生素E琥珀酸酯(D-αtocopherol polyethylene glycol1000succinate, TPGS)与Soluplus存在下的共晶溶出过程中调整给药剂量Cdose(Cdose指该给药剂量浓度下, 药物能够在溶液中达到稳态的平衡浓度值), 探究了Cdose与表面活性剂存在下的共晶平衡溶解度SCC和药物平衡溶解度SD之间的关系。当Cdose>SCC>SD时, 虽然会形成过饱和态, 但可能会由于共晶溶出时表面过饱和度高而发生表面相转变行为, 无溶出优势; 而当SCC>Cdose>SD时, 共晶可以全部溶解形成高过饱和态, 同时当SCC>SD>Cdose与SD>SCC>Cdose时, 由于给药剂量尚达不到平衡溶解度, 无法形成过饱和状态。
此外, 表面活性剂也可通过其他方式对药物共晶溶出产生影响。Ren等选择2%Tween80极大改善了杨梅素−咖啡因共晶的溶出, 且维持过饱和的时间能够达到48h, 推测可能是由于Tween80的使用提升了溶液黏度从而抑制杨梅素结晶成核。表面活性剂也可以通过分子吸附的方式改善共晶溶出, Chen等发现牛磺胆酸钠(sodium taurocholate, STC)对IMC-SAC共晶(001)晶面存在较强的吸附能力, 并通过固有溶出实验证明增大STC浓度会降低配体溶出速率并提升药物溶出速率, 证明了STC会通过对IMC-SAC共晶的吸附作用从而减弱配体从共晶中的“逃逸”速率, 维持药物共晶形式从而改善共晶溶出。
但是, 目前也有研究发现表面活性剂会促进共晶的相转变, 例如在卡马西平−烟酰胺共晶溶出时分别加入0.35mmol·L-1SLS与0.076mmol·L-1 Tween80, 并未起到抑制共晶相转变的作用, 反而加速其转变为卡马西平二水合物, 降低了共晶的稳定性, 并且Tween80还会降低CBZ-NIC共晶的固有溶出速率; 另一方面, 表面活性剂对药物共晶溶出的改善并不代表生物利用度的提高, 因为它不能保证共晶透膜速率的提高, 有可能导致增溶的药物分子被包裹在胶束内部而无法透膜。同时, 表面活性剂的用量也有限制, 过量的表面活性剂也可能会使药物本身的溶解度超过共晶, 导致共晶失去增溶优势。
3.5 添加剂的联用对药物共晶溶出的影响
同时添加适当的高分子聚合物与表面活性剂可以有效提升药物共晶的溶出优势。达那唑−香草醛共晶由于快速相转变, 其体内吸收仅是原料药的1.7倍, 而在处方中加入TPGS作为增溶剂和HPC作为结晶抑制剂后, 共晶的体内吸收提高到了10倍。Li等研究发现FFA-NIC共晶在表面活性剂Soluplus作为增溶剂与聚合物PVPVA作为结晶抑制剂的联用下, 发现通过调整添加剂的使用比例, 可以明显不同程度地改善共晶的溶出行为, 当加入0.2mg·mL-1Soluplus与0.4mg·mL-1PVPVA时, 相较于共晶本身的体外溶出优势可以达到近350倍。目前来看, 添加剂的联用对药物共晶溶出改善有明显作用, 但不同类型添加剂的联合使用研究仍较少, 因此, 探索不同添加剂的联合使用对药物共晶溶出的影响并分析其作用机制对进一步发挥药物共晶溶出优势潜力具有重要意义。
4、难溶性药物共晶体内吸收行为影响因素
4.1 药物共晶渗透性对药物共晶吸收影响
对于BCS分类系统III类与IV类的药物, 药物的渗透性是影响其生物利用度的关键因素, 而药物渗透与药物自身亲脂性、膜扩散性及载体蛋白运输有密切关系。目前有研究已经证明, 药物共晶具有改善药物渗透性的优势, 例如卡马西平−香草酸共晶与卡马西平−琥珀酸共晶对NCM460cells的渗透性相较卡马西平原料药而言均有所改善。共晶配体对药物的增溶作用可能会促进药物的渗透性, Song等成功制备乙酰唑胺−脯氨酸共晶, 在pH1.2、4.0、6.8下, 均明显改善药物溶出, 并通过扩散实验和体内吸收实验, 证明乙酰唑胺−脯氨酸共晶提升了药物的渗透性及生物利用度; Gopi等发现利尿药氢氯噻嗪与不同配体合成的共晶中, 共晶溶解度越高, 其渗透性随之也较高。
也有文献报道抗泌尿系统感染药呋喃妥因与异烟肼等不同配体形成的共晶与物理混合物组对药物透膜的影响差异也较大, 并通过计算模拟手段证明了共晶在溶液中的原子径向分布函数有异于物理混合物, 共晶在溶液中的存在形式可能不同于物理混合物, 从而导致其渗透性不同于物理混合物。
Mannava等指出药物共晶能够改善药物渗透性的原因大致分为两个层面, 一方面药物共晶在溶出时产生药物高过饱和态溶液, 对药物起到了增溶优势, 直接促进了难溶性药物分子的渗透; 另一方面药物共晶可能会降低膜经皮电阻值(transepithelial electrical resistance, TEER), 破坏膜的紧密程度, 从而促进药物分子透膜, 但该情况下可能会导致内毒素进入机体, 这并不是期待的结果, 理想情况应为药物共晶在维持膜完整性的同时改善药物渗透性。
此外, 共晶配体对药物转运的影响也会影响到药物共晶渗透性, Li等研究发现抗病毒药ADD与NIC、SAC与醋氨酚(paracetamol, PA)等配体形成共晶的溶出速率顺序为: ADD-SAC>ADD-PA>ADD-NIC, 但ADD-PA共晶的体内吸收行为优于另外二者, 原因在于PA会抑制P-糖蛋白对药物的外排转运行为, 促进ADD在体吸收, 而另两种配体没有该作用, 所以对药物吸收没有明显优势。
4.2 胃肠道环境对药物共晶吸收影响
目前已上市的药物共晶大部分都以口服途径给药, 因此胃肠道环境也是影响共晶溶出行为和体内吸收的重要因素之一, 推测胃肠道系统主要通过pH变化和生物活性物质等因素影响药物共晶溶出吸收行为。在胃排空过程中, 药物会在胃肠道中经历从酸性到碱性的梯度变化, pH变化范围大约在2~7之间, 这个过程可能会改变共晶组分的电离状态, 进而改变组分的溶解度, 从而进一步影响共晶的溶解度和溶出速率。例如, 中性药物CBZ与弱酸性配体SAC形成的共晶, 由于SAC的溶解度随着pH的增大而增大, 导致共晶的溶解度和溶出速率也随着pH的增大而增大。
除了pH的变化, 胃肠道中还充满了各种生物活性物质, 如磷脂和胆盐等。由于磷脂和胆盐具有特殊的分子结构, 容易组装成胶束具有表面活性, 对难溶性药物的溶解度和生物利用度会产生显著影响。例如, IMC-SAC共晶的相转变行为在STC的存在下得到了显著抑制, 加入STC后的IMC-SAC共晶的体内吸收相较共晶而言改善约1.5倍。
另一方面, 咖啡因是研究较为广泛的共晶配体之一, 咖啡因可能会通过提升胃肠道血流量而改善药物在胃肠道中的吸收行为, 例如Zhu等研究发现黄芩素−咖啡因共晶相对生物利用度为黄芩素的4.1倍, 相对生物利用度的提高一方面归因于共晶溶出速率的提高, 另一方面可能与咖啡因改善胃肠血流有关。
4.3 药物共晶体内吸收行为差异分析
目前大多数共晶体内吸收研究仅停留在现象层面, 涉及药物共晶体内吸收差异机制分析相关研究较少。一般认为, 药物晶体的熔点越低, 晶格能越小, 药物分子的晶体堆积结构越容易解离, 因此推测低熔点共晶可能会促进共晶的溶出与吸收行为, 但许多研究已经发现药物共晶的溶出与吸收行为和共晶熔点/晶格能没有相关关系, 例如难溶性药物柚皮素与不同配体形成的共晶具有不同的体内吸收行为, 柚皮素-L-脯氨酸共晶的大鼠体内吸收是原料药的近5倍, 柚皮素−甜菜碱共晶的大鼠体内吸收仅为原料药的2倍, 其他共晶改善药物吸收的效果更不明显, 而柚皮素-L-脯氨酸共晶熔点为185.58℃, 柚皮素−甜菜碱共晶熔点为124.44℃, 且柚皮素−咖啡因共晶熔点低至96.80℃, 说明低熔点共晶并未呈现出良好的吸收行为。纳米共晶通过联用纳米技术与共晶形式从而达到更加优越的体内吸收行为, 例如黄芩素−烟酰胺纳米共晶的体内吸收为黄芩素约6倍, 而黄芩素−烟酰胺共晶与黄芩素纳米晶的体内吸收仅分别为2.87与3.32倍。Haskins等研究发现槲皮素−己酮可可碱共晶的口服生物利用度相较原料药高64倍, 而槲皮素−茶碱共晶的口服生物利用度仅为原料药7倍, 可见配体的选择对共晶吸收行为也具有重要影响。有研究选取了同类抗菌药物伊曲康唑、泊沙康唑、酮康唑与同一配体4-氨基苯甲酸分别合成共晶, 3种共晶具有体外溶出−体内吸收一致性; 药物的体内吸收行为与药物成核速率、共晶溶出速率及药物在体吸收常数等3种因素具有密切关系, 由于4-氨基苯甲酸具有高渗透性和良好的吸收行为, 该配体不会降低共晶的渗透性从而不会影响药物在体吸收常数, 并且共晶体外溶出环境选择FaSSIF, 与体内吸收环境较为相似, 上述两个因素是3种共晶体外溶出−体内吸收一致的原因所在。药物共晶的体内吸收行为决定药物的生物利用度、药效发挥与用药安全, 需要进一步重点关注和研究。
5、结论与展望
大量研究证明, 药物共晶技术能够提高难溶性药物的溶解度和溶出速率, 但药物共晶在溶出时可能会发生相转变的行为, 导致共晶溶出优势丧失的现象, 因此在共晶制剂研发中, 需要从多方面理解和考虑处方因素从而保证共晶溶出优势的发挥。在共晶体系中引入添加剂是调控共晶溶出的有效方法, 但添加剂影响共晶溶出的相关机制仍有待深入研究。另外, 通过不同添加剂的联合使用, 可使共晶的溶出能够达到较高的过饱和度和较长的维持时间, 更好发挥共晶增溶的潜力。另一方面对于如何更有效选择合适的共晶配体, 从共晶设计角度出发改善难溶性药物理化性质也是值得进一步研究的方向。同时, 应关注添加剂存在下共晶体内吸收的影响因素并通过体内外相关性研究, 从而选择合适的辅料, 优化共晶制剂处方, 更好发挥药物疗效, 推动更多药物共晶产品上市服务于广大患者。
参考文献:《药学学报》2024年

来源:凡默谷


