您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-03-20 07:45
前言:药物晶型是影响药品质量的重要因素,随着药物晶型监管力度逐步增加,单纯的定性分析原料药API或制剂中的晶型已不能满足质量研究的要求,建立多晶型定量分析方法越来越必要。本文主要总结了利用粉末XRD进行多晶型定量分析要点。
X 射线衍射法定量分析的分类
XRPD(X射线粉末衍射法)用于晶型定量,理论基础是Alexander等科学家在1948年推导的衍射峰强度与样品中相应组分晶型含量的数学关系式。X 射线衍射法定量分析可分为全谱图法和单峰法。
全谱图法是以化学计量学为基础,利用最小二乘法(PLS)、全粉末谱图分解或Rietveld法建立全谱峰强度与多晶型组分之间的对应关系。全谱图法不需要标准试样,试样的粒度、择优取向等对定量结果的影响相对也较小,但方法建立非常复杂,对样品图谱的解析要求较高,实际推广应用较少。
单峰法是建立特征峰强度与被测药物中某一晶型组分之间的对应关系。单峰法定量分析模型简单、操作快速,但仪器参数、试样粒度、择优取向等均会影响分析结果的准确度,需要严格控制操作条件才能得到可靠的数据。相较而言,单峰法实际应用更广。
以实际推广应用为主,以下总结XRD单峰法多晶型定量分析过程中操作条件的控制以及参数的影响。
XRD单峰法定量分析模型建立
分析模型建立:单峰法晶型定量分析本质是建立特征峰衍射强度与晶型含量之间的线性关系。单峰法用于定量的数学关系式为:
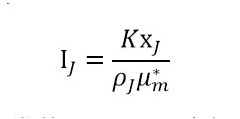
式中,为组分J的衍射峰强度;K为常数,m-1;XJ 为组分J的质量分数;ρJ 为组分J的密度,kg/m3;为各晶型组分的质量吸收系数,m2/kg。μm药物分子的不同晶型的质量吸收系数较小且其分布范围也较小,药物晶型定量研究中,可假设同一物质不同晶型的质量吸收系数相等。因此衍射峰强度与晶型含量之间可简化为线性关系:IJ =KXJ 。简而言之,原料药或制剂中晶型的定量分析模型建立,即建立如下图所示的标准曲线和验证曲线。
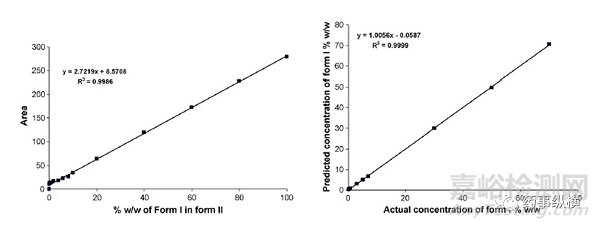
图1定量分析的标准曲线和验证曲线
模型的评价标准:定量限和检测限
ICHQ2(R1)分析方法论证中定量限和检测限的定义,迁移至XRD定量分析中,含义分别如下:XRD的定量限(LOQ),是指在合适的准确性和精密度下,能够定量测定样品中被分析晶体的最低量;XRD的检测限(LOD)是指样品中的被分析晶型能够被检测到的最低量,但不一定要准确定量。
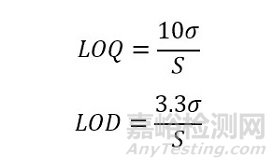
σ是指实测值与标准曲线预测值的标准方差,S是指校准曲线的斜率。
XRD单峰法定量分析准确度的影响因素
XRD单峰法定量分析准确度依赖于XRD衍射峰强度的准确测量, XRD衍射峰强度受多种因素的影响。Mier在文献中总结影响衍射峰强度的准确测定的参数有33项。结合药物晶型研发经验,药物晶型定量分析建模过程中对应需要优化的主要参数如下:
(1)XRD仪器参数的优化
XRD广泛应用于材料领域,检测石英和检测药物晶体的参数设置如电流、电压、发射狭缝、接收狭缝等天差万别。用于药物晶型研究的XRD基本设置一般都比较固定。在药物定量分析中,主要通过“步长”和“停留时间”优化扫描速度,以期平衡分辨率(分峰质量)及扫描时间。因为扫描速度越慢,峰的分辨率越高,但对应检测时间越长:Scan rate = Step Size/Steptime。图2是奥氮平定量分析中扫描速度的优化,选择扫描速度0.6°/min(D方案),特征峰检查率最高且用时较短。
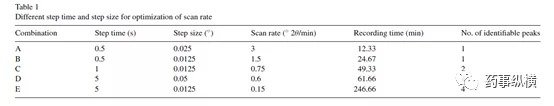
图2 奥氮平(FormI:FormII=5%)不同扫描速度的特征峰检出个数
这里顺带提一下,评价定量分析模型的标准是定量限和检测限,定量分析的难点也在于低含量组分的检出限。因此优化设备参数、特征峰的选择以及样品制备方法的优化均以低含量晶型来考察和优化。
(2)样品制备参数的选择
样品粒度:研究表明,样品的择优取向对衍射峰强度影响最大(可导致样品衍射峰强度的变化至100%),而样品充分研磨过筛是最有效减少样品择优取向的影响。一般研磨样品过360目筛子的粉末用于定量分析。但对于静电原因无法深度研磨过筛或者研磨会导致样品转晶,则需要考察不同筛子级别的粉末样品多次测试(一般测六次)衍射峰强度的波动性,选择衍射峰强度波动最小的粒径用于定量分析。
装样量:定量分析中,每一个分析样品均为准确称量,如所有装样量均为500mg或者300mg等,固定装样量可以排除样品量过少对峰强度的影响,也可最大限度减少批次“试样平面”影响。
标准样品的配置:定量分析标准曲线样品配置,主要原则是低含量数据多,因为低含量组成是定量分析误差主要来源。例如在奥氮平晶型定量模型中,标准样品的配置方案如下:Form I(被定量组分)在混晶中的质量含量:0、0.1、0.2、0.4、0.6、0.8、1、2、4、6、8、10、20、40、60、80、100。
样品检测的重现性:ICHQ2(R1)分析方法论证中定义,样品的重现性是指不同实验之间的精密度。在XRD定量分析中,考察重现性的方法汇总如下表:
表1样品检测重现性考察表

(3)特征峰的选择
晶型定量特征峰选择的依据:1,独有的峰,与其他晶型的衍射峰无交叉;2-满足1的条件下尽可能的选择峰强高的峰。如图3所示,奥氮平Form I、II的定量分析中,Form I是低含量晶型,Form II是主晶,定量模型建立中,Form I强度最高的峰与Form II峰存在叠加,不选择其作为特征峰。其他特征峰中(*标注)中2θ在18.48°,相对峰强最高(I/I0=78.8%),因此选择18.48°处的特征峰作为定量分析的特征峰。
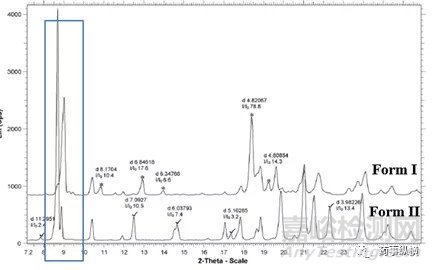
图3 奥氮平Form I(*标注)和Form II(√标注)特征峰
在《药物晶体粉末XRD图谱分析》讨论中,有留言问,特征峰选择几个比较靠谱?这个具体问题具体分析。
定性分析中,一般会将相对峰强度≥10%的峰均列为特征峰,而对于需要着重强调晶型区分度的情况下,2θ角度不同的峰(尤其在低角度)不论相对峰强度如何,均需要确定为特征峰。
而定量分析中,如果单个特征峰建立的分析方法满足要求的定量限和检测限,选择一个最优特征峰进行定量分析即可(简单省事)。但对于单个特征峰不能满足的,可以选择多个,通过建立特征峰强度比,面积比等建立线性关系。如下图4为API多个特征峰分析建模的比较,2θ=22°峰高与晶型含量建立线性关系,或者2θ=22°峰强度/18-30°峰强度总和与晶型含量建立标准曲线,检测限和定量限相对较高。
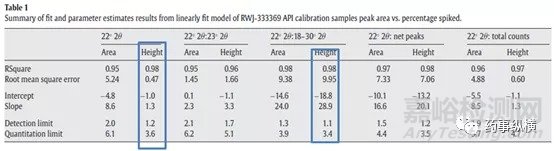
图4 不同的特征峰参数与晶型含量线性关系的比较
小结
目前XRPD单峰法仍是多晶型定量的主要手段,但样品制备过程以及晶体粒度、晶习等带来择优取向会严重影响定量分析的准确度,XRD定量分析方法的开发显得尤为重要。此外光谱法(近红外、拉曼、衰减全反射红外光谱法ATR-IR)和热分析法(DSC)等在晶型定量分析中也越来越多的被应用,很难说哪一种晶型定性方法更好,不管怎么样,选择一种合适的晶型定量分析方法需综合考虑方法的难易程度、灵敏性、耐用性、准确性和试样的特性等方面。
参考文献
[1] 肖燕, 王静康, 尹秋响,等. 固体药物晶型定量分析方法[J]. 石油化工, 2015, 44(1):11-18.
[2] Tiwari M , Chawla G , Bansal A K . Quantification of olanzapinepolymorphs using powder X-ray diffraction technique.[J]. Journal ofPharmaceutical & Biomedical Analysis, 2007, 43(3):865-872.
[3] 《中国药典》2015年版新增"药品晶型研究及晶型质量控制指导原则"简介
[4]Varasteh M , Deng Z ,Hwang H , et al. Quantitative determination of polymorphic impurity by X-raypowder diffractometry in an OROS formulation.[J]. International Journal ofPharmaceutics, 2009, 366(1-2):74-81.
[5]Mier J , Leon L D , Castillo A , et al. Accurate quantification of quartz andother phases by powder X-ray diffractometry[J]. Analytica Chimica Acta, 1997,337(3).
[6] ICH Q2(R1)分析方法论证:正文和方法学

来源:药事纵横


