您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2019-04-17 10:04
厂商如果开展CE认证,通常需要准备两部分资料,一部分是体系文件,另一部分是技术文件。其中,技术文件是证明产品安全性和有效性最直接有力的证据。所以,准备一套完整并符合法规要求的技术文件就变得至关重要了。相比于CE MDD指令,新版CE MDR法规为技术文件单独设置了附录2,对其提出了更明确的要求。小编今天就和大家好好讨论一下这份技术文件清单。
CE MDR技术文件清单
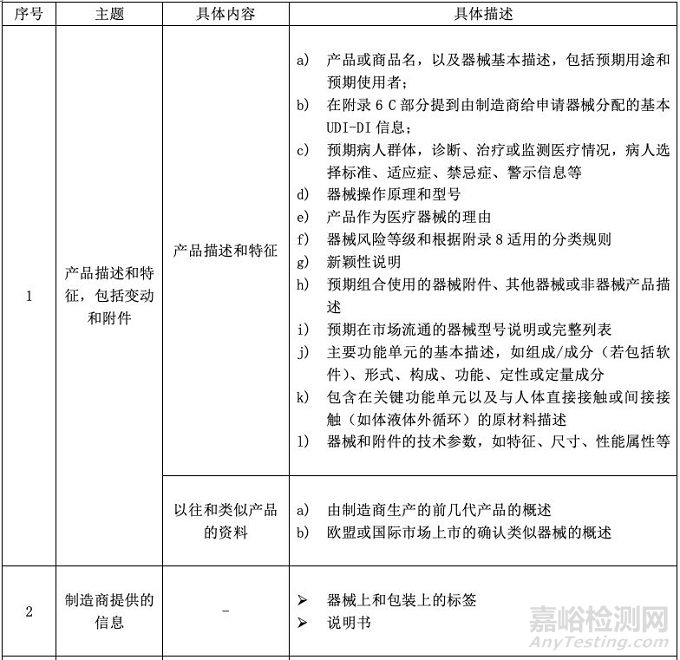
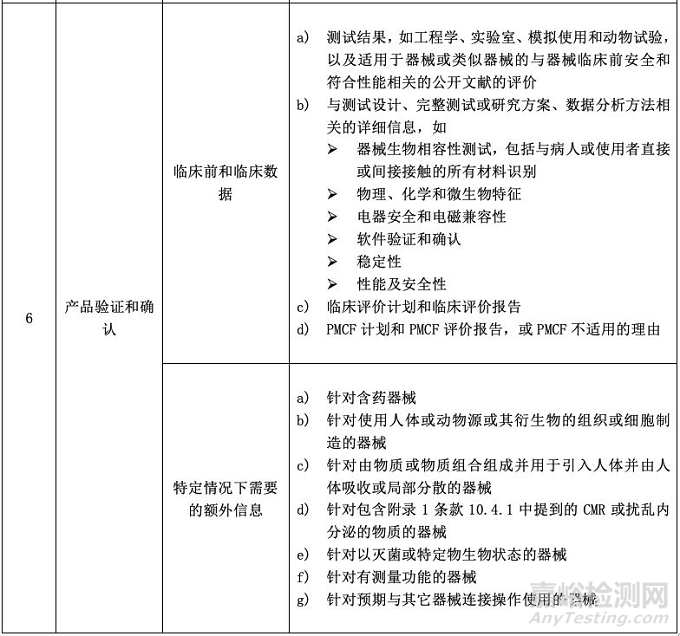
CE MDR附录2将技术文件分成了6大块,并要求技术文件和相关总结应该以一种清晰的、有组织的、可检索的且非模棱两可的形式呈现,同时应包含附件2中涉及的内容。小编将具体内容整理成表格形式,供大家参考。

总结
通过上述表格的介绍,大家对于CE技术文件要包含的内容应该有了大概了解。针对各部分的关键点,小编再做一些简单总结:
关于第一部分“产品描述和特征”,小编认为万变不离其宗,熟知产品是关键,厂商要能够将产品的来龙去脉一一道明,同时提供相应的资料;
在第二部分“制造商提供的信息”中,重点是满足标签和说明书的标准要求,即EN ISO 15223-1:2016和EN 1041:2008;
关于第三部分“设计和制造信息”,厂商应该对产品的设计和制造过程了然于胸,通过提供生产工艺图、过程检和成品检等信息,让审核员能够清晰地了解“产品从何而来”以及“产品质量是否有保障”;
在第四部分“基本安全和性能要求”中,厂商在填写检查表时通常要遵循三步法,即罗列产品满足的最新标准,明确适用的条款要求,提供符合性证据或不满足条款的说明;
关于第五部分“收益风险分析和风险管理”,厂商要严格按照标准EN ISO 14971:2012的要求编写风险管理计划和报告,同时要特别关注欧盟对于风险管理的特殊要求(在标准ANNEX ZA-ANNEX ZC部分) ,比如“As far as possible原则”、“披露剩余风险不算是风险控制措施”等差异;
在最后一部分“产品验证和确认”中,厂商要基于产品要求,按照最新的相关标准开展相应测试,并理清测试报告的相互关系,提供具有逻辑性的证据;同时,临床评价应按照MEDDEV 2.7.1/Rev.4开展,并根据产品类别及风险决定是否开展PMCF;至于特定情况下需要的额外信息,则要根据产品特性来判定是否需要提供。

来源:启升资讯


