您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2020-06-24 16:10
医疗器械未知可沥滤物评价方法建立及表征技术审查指导原则
一.前言
对医疗器械进行充分的化学表征是医疗器械产品设计开发环节中重要内容,可以为材料的选择、工艺优化等提供证据,同时医疗器械的化学表征提供了医疗器械生物学评价和毒理学风险评估所必须的信息,除此之外,化学表征还可以:
1.支持再处理医疗器械的生物学安全性;
2.临床使用条件下可沥滤物的识别和量的确定可用于支持毒理学风险评估;
3.在临床接触性质相同时,支持医疗器械与市售器械的等同性;
4.支持生产工艺(如灭菌工艺、清洁工艺等)、生产地址、材料或组件、供应商等发生变更时,与市售器械的等同性;
5.支持拟用材料与市售器械/材料在材料组成或浸出物谱(extractable profile)方面的等同性;
6.支持器械终产品与原型器械(prototype device)的等同性评估;
7.针对器械的临床预期应用,支持新材料的筛选等。
需要说明的是,单纯的化学表征一般不足以证明器械/材料的等同性,
也不足以确定器械/材料的生物相容性,需要由具备专门知识且经验丰富的人评估其充分性,必要时需要结合毒理学资料、器械/材料的物理学特征、后续处理、临床用途等进行综合评估。
医疗器械的化学表征一般包括确定产品组分成分研究(如产品结构、成分、理化特性等),浸提研究(extractable study)、可沥滤物研究(leachable study)等,可沥滤物研究是医疗器械化学表征的重要内容。医疗器械可沥滤物(Leachables)是指医疗器械或材料在临床使用过程中释放出的物质的统称,一般包括灭菌残留剂、工艺残留物、降解产物以及材料中的单体及添加剂(包括稳定剂、抗氧化剂、增塑剂、着色剂等)等。作为医疗器械生物学评价的一部分,需要评估医疗器械产品在与人体直接或间接接触并发挥作用的过程中,可沥滤物对人体安全性方面的潜在风险。根据可沥滤物研究体系不同,分为根据相关信息识别的已知可沥滤物(Target Leachables)和根据未知可沥滤物研究体系鉴别的未知可沥滤物(Unspecified or Unknown Leachables)。本指南即提供了未知可沥滤物研究体系的一般要求。
随着现代分析技术及评价理念的进步和完善,包括风险管理下医疗器
械生物学评价理念的不断完善,以未知可沥滤物评价体系为基础的可沥滤物研究表征体系已成为医疗器械化学表征的重要组成部分,从而在医疗器械生物学评价等前述应用中,发挥着不可或缺的重要作用。
本指导原则是对医疗器械未知可沥滤物研究评价方法及表征技术的一般要求,申请者应依据具体产品的特性和研究目的对注册申报资料的内容进行充实和细化,并对在研究过程中评价技术的设计、实施、结果的应用的科学性和合理性进行充分的阐述。
本指导原则旨在帮助和指导申请者对医疗器械产品注册申报资料进行准备,以满足技术审评的基本要求。同时有助于审评机构对该类产品进行科学规范的审评,提高审评工作的质量和效率。
本指导原则是对申请者和审查人员的指导性文件, 但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其它方法,也可以采用,但是需要提供详细的研究资料和验证资料。应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将适时的进行调整。
二.适用范围
本指导原则适用于医疗器械注册申报或产品开发等环节时,针对不同评价目的,在进行未知可沥滤物研究时,对医疗器械未知可沥滤物评价方法建立和表征提供参考。
三.未知可沥滤物表征研究方法及一般步骤
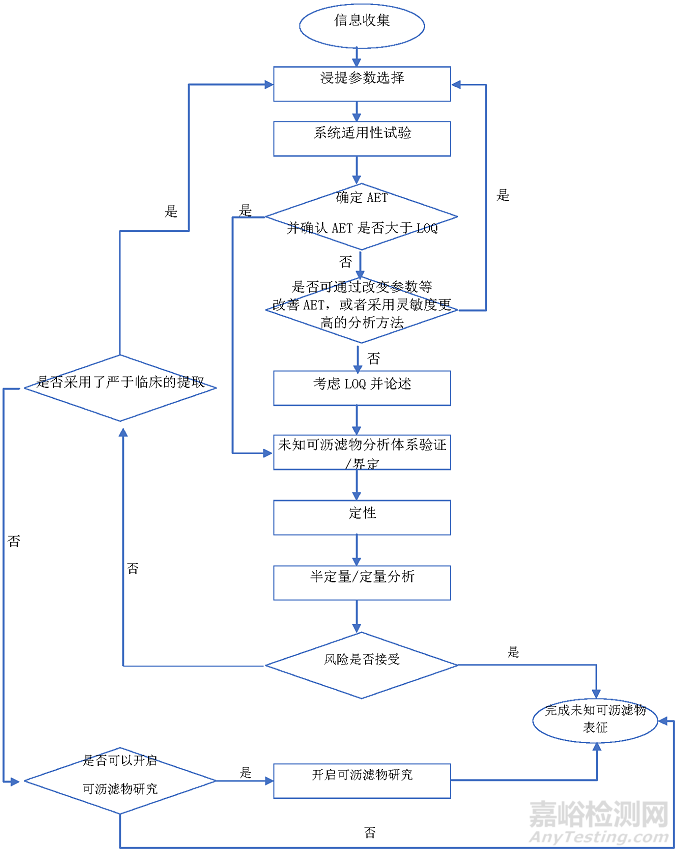
作为医疗器械生物学评价的一部分,尤其是作为医疗器械毒理学风险评估必不可少的研究步骤,未知可沥滤物的研究程序必将与毒理学风险评估存在着不可分割的联系。因此,结合风险评估的要求,未知可沥滤物研究的一般程序如下:
首先,是进行拟研究器械与未知可沥滤物表征相关的充分的信息收集;其次,根据所研究器械的临床使用特性,论述并选择适宜的浸提参数。由于相当数量的医疗器械临床使用特点决定了无法开展临床使用条件下的可沥滤物研究,因而浸提参数的选择和论述尤为重要。在此基础上,应采用适宜的混合参考物和内标开展系统适用性试验和评估。
在上述所收集信息,尤其是临床使用信息,结合所选择的浸提参数,尤其是浸提比例等参数的基础上,计算和确定分析评估阈值(AET),并确认AET值是否高于分析方法的定量限(LOQ)值。当AET低于分析方法的LOQ时,可以考虑是否可以通过改变浸提参数提高AET或者采用灵敏度更高的分析方法,从而确保分析体系的LOQ值能够满足AET的使用要求。如果通过上述方法改变仍然无法获得AET高于LOQ的结果,则可能需要考虑论述并使用所对应分析方法的LOQ代替AET,并给出合理的依据。
完成AET确认(或最终不得不采用LOQ代替AET),后续的研究则进入到未知可沥滤物分析体系的建立和验证过程,以及随之完成的定性、半定量/定量研究。通过毒理学风险评估判定来自器械的可沥滤物风险是否可接受。若风险可接受,则结束未知可沥滤物表征。在毒理学风险评估过程中若发现风险不可接受,则考虑浸提参数的选择是否采用了严于临床的浸提过程,若是,则可以通过改变浸提参数,使得浸提参数更加接近于临床实际,重新确认浸提参数,同时,重新进行上述研究步骤。
若已采用了接近于临床使用的浸提参数,采用替代溶剂获得的可浸提物研究结果不能支持等同性,或者通过毒理学风险评估,判定此结果不能接受,则可以考虑器械是否具有开展实际接触条件下(如血液接触)的可沥滤物研究的可行性。若可行,可进一步开展实际接触介质的可沥滤物研究,并结束未知可沥滤物表征,形成报告用于进一步的毒理学风险评估。若器械临床接触性质决定不能开展实际接触介质的可沥滤物研究,如某些组织接触的植入物,则可以考虑开展经论述的、模拟生理环境的未知可沥滤物表征的可行性,完成未知可沥滤物表征,形成报告用于进一步的毒理学风险评估。
对各环节的论述及注意事项可参考附录A流程图并参考后续讨论。
(一) 信息收集
充分的信息搜集能有助于产品的可沥滤物研究分析,可以为后续未知可沥滤物的定性提供更精确的信息,因此进行生物学评价特别是化学表征的首要步骤是进行充分的信息收集。信息的收集可包括器械的结构及材料组成、来自原材料供应商的信息、拟研究材料/器械的理化特性及与可沥滤物相关的文献信息、器械的生产工艺信息、已有的历史数据库信息、临床应用信息等。
上述信息主要预期用于未知可沥滤物实验研究,并不一定作为全部递交资料内容。况且上述信息在实际收集过程中也不一定全部具有可获性。信息收集的主要目的仍然是尽可能地服务于未知可沥滤物的表征。
具体考虑的信息如下:
1)器械的结构及材料组成信息:包括构成拟申报器械的组件或成分名称、每种组件或成分的材料名称、材料或成分的CAS号、材料供应商来源及牌号(若有)、每个组件或成分是否与人体接触、以何种形式接触(如,直接、间接或不接触等)。
2)来自于原材料供应商的信息:包括每种原材料添加剂名称、添加剂的CAS号、每种添加剂的组成比例(若可行)、原材料工艺信息和加工助剂信息(若可行)、材料分析证书等。
3)拟研究材料/器械的理化特性及与可沥滤物相关的文献信息:包括但不限于材料/器械的基本理化特性,如溶解特性、是否为可吸收材料/可降解的材料/器械,若为可降解材料/器械,来自文献或已有数据库中确认的降解机理、降解产物或相互作用产物。还包括与材料或器械相关的来自文献或历史数据库的可沥滤物信息等。
4)器械的生产工艺信息:包括材料/器械的生产工艺流程图、关键参数(如温度)、加工助剂(若有,如清洗剂、润滑剂、脱模剂等)名称及CAS号,若为灭菌产品,灭菌方式及关键参数(若可行)等。
5)已有的历史数据库信息:该部分信息是基于风险评估理念减少不必要的实验研究和增加研究可靠性和可信性的重要手段。材料/器械的历史数据库信息除可能包含以上所描述信息外,结合拟申报器械的临床应用性质,来自于材料/器械的已知可沥滤物、未知可沥滤物、降解特性及降解产物、反应产物及中间产物等的实验研究结果,包括研究方法所形成的数据库,是开展拟申报材料/器械未知可沥滤物研究的重要输入。甚至是在某些情况下,经过充分评估,有可能成为免除新的可沥滤物研究的重要依据。
6)与未知可沥滤物研究相关的临床应用信息包括:临床接触途径、接触时间及频率、应用人群、临床使用方式:如,参照说明书或其他相关文件获得的每天最大使用量信息、使用前的处理方式(若适用),如清洗、预冲、预加热、预混等、是否在位聚合等特殊使用信息、若为可吸收产品,预期的体内降解时间等。
上述1)~5)信息收集的结果可用于:1)在材料/器械未知可沥滤物研究体系建立过程中帮助选择适宜的相关参考品,用于分析体系的系统适用性开发、建立,以及方法学的界定(method qualification)等;2)在对浸提物谱(extractable profile)/可沥滤物谱(leachable profile)进行未知可沥滤物鉴别时,上述信息的获得也是对未知可沥滤物鉴别及最终确认的重要支持证据之一;3)前述信息也是对未知可沥滤物来源进行解释的重要依据之一。而6)的信息收集是建立分析评估阈值(AET)、确定浸提方式的必须要素,如考虑采用极限浸提、加严浸提、模拟浸提,包括模拟参数等。另外,在通过半定量(semi-quantitative analysis)或定量分析(quantitative analysis)获得的可沥滤物进行毒理学风险评估时,也是毒理学家在评估过程中所需的必要信息。
需要说明的是,某些情况下1)~5)的信息可能由于各种原因不能全部获得,由于上述信息收集的目的是有助于未知可沥滤物表征,如果在实际未知可沥滤物研究过程中根据需要进一步获得材料成分信息,可以参照本指导原则建立的未知可沥滤物体系进行研究以获得所需信息。所不同的是,这种以材料组成信息为目的的浸提研究一般采取消解、溶解或极限浸提方式,但这种以材料表征为目的可浸提物研究不应视为信息收集的必须步骤。
由于未知可沥滤物研究一般会采用毒理学关注阈值TTC(相关标准参见ISO/TS21726)来导出分析评估阈值(AET)并应用于未知可沥滤物评价体系,因而,在信息收集阶段,基于已获得的信息,某些情况下可能需要考虑是否存在ISO/TS21726所提到的特殊关注化学物质的可能性。
(二)建立分析评估阈值(AET)
未知可沥滤物和可浸提物分析过程是采用一种旨在发现、鉴定和半定量的筛选分析方法进行分析,最终形成可浸提物谱(extractable profile)和可沥滤物谱(leachable profile)的一个过程。由于可浸提物和/或可沥滤物的多样性,以及分析方法的灵敏性等原因,以及毒理学关注阈值的考虑,不一定需要对所有的物质均进行定性定量分析。因此对于未知可沥滤分析的首要任务就是建立分析评价阈值(AET),当未知可沥滤物的量高于该阈值时,需要对其进行定性和定量分析以进行充分的毒理学风险评价;当未知可沥滤物的量低于该分析评价阈值时,则可以在不知晓其化学结构和毒理学信息的情况下默认其毒理学风险可接受。
AET的建立一般是根据毒理学关注阈值(TTC)或安全阈值(SCT),并充分考虑用于浸提的产品数量、浸提液体积以及基于筛选方法的分析不确定因子等因素换算后得到,具体计算公式如下:
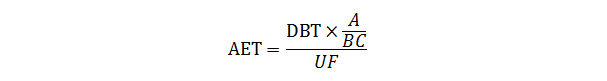
其中:DBT ——基于剂量的阈值,如采用TTC或SCT
A——浸提研究使用的器械数量
B——浸提溶剂体积
C——临床每天使用器械数量
UF——不确定因子
其中,需要注意的是:
1) 采用TTC计算AET时,TTC的数值可参照ISO/TS21726,但当信息收集过程中表明器械或材料中含有ISO/TS21726所提到的特殊关注化学物质,AET并不适用于该类物质的评估。
2) 尽管已确定单一金属的允许日接触量(PDE),但尚未确定适用于所有金属的基于剂量的阈值(DBT)。因此,实际上,AET仅适用于有机可浸提物或可沥滤物。
3) 为了保证毒理学风险评估更全面、准确、有效,确保分析方法能够满足毒理学风险评估的要求,分析方法的定量限(LOQ)必须低于AET。但在某些情况下,当系统灵敏度不能满足AET要求时,也可以采用LOQ作为AET。如一些体积较大的医疗器械采用模拟浸提时。但是毒理评估中要考虑AET和LOQ的差异,并对差异合理性进行论述。
4) 不确定性因子UF的选择及建立,可参考未知可沥滤物分析体系部分中系统适用性论述。
(5)当研究发现产品中可能存在前述特殊关注物质时,应首先考虑能否
直接更改产生该物质的材料或对现有工艺进行优化以将其含量降至最低,并开发高灵敏度的专属的并满足更低的检测限和定量限的检测方法对其进行检测及安全性评估。
(三)浸提条件选择
基于风险评估的需要,进行未知可沥滤物研究时需要确定假设的最不利情况下的化学释放,以评估其最恶劣环境下的可能风险。例如,在临床使用过程中,器械的全部成分都被个体吸收,则视为医疗器械产生的化学影响达到最高程度。例如,某些植入式医疗器械预期在临床使用过程中溶解,或者外部接入器械在临床使用过程中可沥滤物的完全析出,还应考虑其它因素,例如医疗器械的体积等。除此之外,假设的最不利情况还包括产品风险最大的规格型号、最长使用时间、接触介质等,以便重现可沥滤物最大人体暴露量相关的使用条件。
1.概述
浸提旨在获取等于或超过临床使用中产生的可沥滤物的可浸提物谱,但又不会对材料或可浸提物谱产生不良影响(例如,材料的降解、可浸提物的化学改变等)。这样做可提供至少与医疗器械的可沥滤物谱同样广泛的浸提物谱,即可浸提物谱应尽可能全面反映可沥滤物及其水平。然而,应注意某些情况下并非所有可沥滤物均必然存在于浸提物谱中,例如可降解产品等。
另外,采用了与模拟使用条件有较大差异的浸提溶剂、浸提方法时,所获得的可浸提物可能很难完全反映出模拟使用条件下获得的每种可沥滤物特征。
通常情况下,用于化学表征的浸提研究的应用包含以下四个方面:
1)用于生成医疗器械构造或制造材料的化学信息;
2)用于生成医疗器械或材料最坏情况下的可浸提物谱;
3)用于生成医疗器械或材料在临床使用条件下(模拟浸提)的可浸提物谱;
4)用于化学表征数据与ISO 10993中所述的生物学试验结果关联性分析。
一般来说,浸提都是一个复杂过程,受时间、温度、表面积与体积比、浸提介质和相对于浸提介质的供试品中物质分配行为等方面的影响,同时可能还包括考虑材料自身的化学特性。通常,浸提条件不应改变供试品,因为改变供试品可能会导致供试品所释放的可浸提物数量和/或类型发生变化。总的来说,为了确保风险评估的充分性,可浸提物研究应至少考虑以下原则:
1)未知可沥滤物研究的分析方法应是基于扫描技术来完成的,因而应选择高灵敏度、高选择性的分析方法进行,以尽可能的识别所有潜在的可沥滤物,并对高于AET的物质进行定性定量分析。因此分析方法体系应确保能检测到所有潜在的浸提物,且能够为浸提物鉴别提供足够的信息,同时可进行半定量或定量分析。
2)如果采用加严浸提或极限浸提,应对浸提条件进行论述,以确保浸提条件不会对浸提物产生影响,从而导致研究结果与临床实际接触情况相关性差。
2.浸提及参数的论述
浸提的主要目标是获得至少与器械可沥滤物谱一样全面的可浸提物谱,这意味着可浸提物谱应尽可能包含所有可沥滤物,并且浓度不低于可沥滤物浓度。但必须限制过高估计的程度,例如,过高估计可浸提物浓度,会增加毒理学风险评定中的不确定性,过严的浸提条件也可能会导致可浸提物谱发生改变并产生降解产物。一般来说,浸提条件的选择常需要考虑以下因素:
2.1 浸提方式。常用的浸提方式包括模拟浸提、加严浸提、加速浸提和极限浸提(具体可参考GB/T 16886.12和GB/T16886.18中给出的定义)。由于浸提物研究的主要目的是建立医疗器械或材料的最坏或临床使用条件下的可浸提物谱,因此推荐的浸提方式是严于临床的加严浸提或极限浸提,对于某些在实验室内可实现临床条件重现的器械,可选择模拟浸提。
极限浸提可建立从医疗器械或材料中浸提的最大可浸提物的量,即医疗器械或材料在临床使用/寿命期间可能释放的可沥滤物量的最大量。加严浸提是指在加严浸提条件(在一个或多个维度方面,相对于临床使用条件)完成的浸提。例如,考虑到以下一种或多种情况:1)浸提温度超过临床使用温度;2)浸提时间超过临床使用时间;3)介质的浸提能力超过临床接触溶液的浸提能力;4)器械表面积与浸提溶剂体积比超过临床使用接触量。为了保证毒理学风险评估的充分性,所采用浸提条件应至少与临床使用条件一样。
一般来说,加严浸提可用于短期或长期接触医疗器械的分析。由于持久接触和很多长期接触类器械,很难模拟其临床使用条件,对于该类器械一般选择极限浸提,论证需充分考虑到介质的pH、极性、浸提温度、比表面积等因素的影响。当极限浸提和加严浸提结果不能满足毒理要求时,可采用以时间设计为目的的浸提动力学研究,如测定每日释放量,绘制趋势图。
对于需要进行未知可沥滤物研究的器械,在未知可沥滤物表征阶段,也可采用极限浸提,以获得尽可能多的信息,同时便于初步的毒理学风险评估。若风险可接受,则不必进行加严和模拟浸提。如果极限浸提结果不能满足毒理学要求,可考虑加严浸提或模拟浸提。同时也可以直接选择模拟浸提或加严浸提,一般情况下模拟浸提是通过使用模拟临床使用条件的浸提条件(即温度和持续时间)使用合适的浸提溶剂来完成浸提。如果选择使用替代溶剂进行模拟浸提,应论述替代溶剂的合理性。
对可降解医疗器械某些情况下,浸提过程可能不能体现临床使用过程中出现的降解产物或其他副产物,因而宜采用其他途径获得该类信息,如文献研究、体内/体外降解研究。一般情况下,宜优先考虑文献研究,而对于大多数材料或器械来说,可能充分的文献研究即可完成该类信息的输入。上述研究结果,需要时,可在毒理学风险评估过程中一并纳入考虑。
2.2 浸提的重复次数。大多数情况下每种浸提溶剂一般需要进行两平行浸提,但是对于一些可变性较高的器械或材料,如原位聚合的器械和可吸收器械,可能需要三平行浸提。如果在进行平行制备时发现浸出物谱有较大变异性,尤其是化合物数量的显著不同,宜首先考虑分析系统的原因调查,以对是否需要增加浸提次数进行确认。
2.3 浸提溶剂选择。浸提溶剂的选择是保障化学表征充分的重要因素,在选择浸提介质时,应考虑器械或材料在临床接触介质的性质,比如酸碱性,极性等。浸提溶剂首先不应引起测试器械或材料的改变,因为这种变化可能会导致释放的可浸提物的量和/或类型的改变。也不应该影响浸提物谱,如果存在这种情况应在报告中给予论述,如使用醇类浸提溶剂时可能考虑会与有机酸类浸提物发生酯化反应的可能性。
浸提溶剂的选择可能涉及一种或多种浸提溶剂,由于未知可沥滤物的不确定性,某些情况下,单一浸提溶剂可能并不能保证可浸提物谱能有效涵盖可沥滤物谱。例如血液接触类器械一般选择极性和中等极性溶剂,对于长期及持久接触植入物,一般使用极性、中等极性和非极性三种不同极性溶剂。同时对于某些间接接触医疗器械,可以使用单个浸提溶剂来重现预期的接触液体,例如充填氯化钠注射液的预充式导管冲洗器,可选择使用充填的氯化钠注射液作为浸提溶剂。对于金属元素的浸提研究时,可选择使用生理盐水或模拟人体组织液的弱酸溶液作为浸提溶剂,必要时还需要进行有机溶剂的浸提研究,例如某些含有有机金属催化剂的器械。
无论选择何种浸提溶剂,都应对浸提溶剂的选择过程进行论述。另外,溶剂的选择可能综合考虑器械临床接触性质、溶剂与器械的兼容性和溶剂与分析方法的兼容性等因素。关于溶剂与器械的兼容性,已在“2.3浸提溶剂的选择”中描述。关于溶剂与分析方法的兼容性,主要考虑与分析方法所使用仪器的兼容性,如实践当中,对中等极性性溶剂的选择通常以醇类为主,兼顾考虑了LC和GC方法的兼容性。又如,采用缓冲液或水做溶剂进行GC/MS分析时,如果采取直接进样方式,会因为水的蒸气体积,盐类效应等影响分析结果及色谱设备。所以,一般会进行液液萃取将缓冲液或水中的有机化合物转移到合适的有机溶剂比如二氯甲烷或正己烷当中,再进行气相-质谱分析。”
2.4浸提技术。提取方式可分为“传统”和“现代”两大类。通用的传统浸提技术包括索氏提取、超声提取、震荡提取和回流提取等;现代的浸提技术包括微波辅助萃取、流体萃取、超临界流体萃取等。由于现代技术一般用于已知物的处理和制备,并没有很多的验证用于未知可沥滤物研究,例如,某些情况下可能会造成可沥滤物谱发生较大改变而产生分析假象,因而现代技术的应用需要经过合理的论述或验证。各种浸提技术都具有各自的优点和局限,如回流的效率较高,但提取介质为水溶液时,由于水的沸点较高,回流则过于苛刻,可能导致某些有机可提取物发生进一步的降解。索氏提取虽然可避免溶剂沸点带来的分析假象,但由于较大的溶剂体积可能为分析体系灵敏度带来一定挑战。由于“传统”浸提技术经济可行且易于操作,同时“传统”浸提技术应用时间久,各种浸提技术的功能和性能是众所周知的,并且有充分的记录,因此相对而言“传统”浸提技术应用更为普遍。
无论采取任何“传统”或“现代”浸提技术,申请人都应充分考虑其技术和实际限制,以及与医疗器械临床应用的相关性及兼容性。浸提过程不应有影响评价的额外引入物质甚至干扰最后的安全性评价,应通过空白对照试验等方式确认。
除此之外,某些情况下可能需要进行释放动力学研究,例如某些植入式可降解材料,因可能存在化学物质不均匀释放,应考虑特定降解时间点未知可沥滤物峰值释放等极端条件。再比如在上述浸提参数设计中,采用极限浸提或加严的浸提,尤其是采用“(三)浸提条件选择中2.1”所述浸提技术获得的可浸提物不符合毒理学风险评估要求时,则可能会模拟更加接近实际使用的浸提技术,如采用37℃,并选择便于进行毒理学评估的适宜的取样时间点,获得浸提曲线。这种情况下的浸提研究可能会需要更长的浸提时间。
3.浸提液的制备(必要时)
为了尽可能防止可浸提物的丢失,原则上应尽可能对浸提液直接分析,尽可能不采用其它方式对浸提液进行处理。
但某些情况下可能需要对浸提液进行一定的制备才能进入分析程序。例如由于浸提溶剂与仪器的兼容性问题,可能需要对浸提液进行转换才能实现分析。需要时,可在溶剂转换过程中同时完成浸提液的浓缩。由于溶剂转换容易造成可沥滤物/浸提物的损失,因而一般需要通过加标回收实验,即通过加入不同极性及离子化物质等验证溶剂转换的可靠性。再比如,有些情况下,衍生化可能有助于某些未知可沥滤物的识别,这种情况下就需要注意衍生化反应的充分性以及可能带来的干扰等问题。而对于某些器械,如动物源或植物源可吸收产品获得的浸提液有可能存在浑浊等现象,在确认浑浊等现象的基础上,可采取离心、过滤、溶剂转换等处理方式。一般需要对收集到的不溶性物质进行化学表征,如红外表征,或适宜溶剂复溶后的定性定量,同时论述不溶性物质的来源。无论采用何种处理方式,均应充分的记录和阐述。某些特殊情况下,对于复杂基质可能需要溶剂转换实现未知可沥滤物扫描。
(四)未知可沥滤物分析体系的建立
未知可沥滤物表征,与已知可沥滤物(目标可沥滤物)测定的最大不同,在于建立一套可靠的未知可沥滤物分析体系。该分析体系为多种分析技术联合体系,并采用未知可沥滤物扫描模式实现未知可沥滤物发现的过程。分析体系应该在现有技术水平上,符合毒理学风险评估原则下,保证对浸提物的可靠发现、鉴别和定量,即分析体系应保证具有足够的灵敏度和分离各种化学结构的潜在可沥滤物的能力,同时完成未知可沥滤物的半定量。
未知可沥滤物分析体系的建立需要考虑系统适用性、方法学考察、定性和定量研究等方面,其中系统适用性是开发建立未知可沥滤物分析体系的重点。
1. 系统适用性
系统适用性试验主要是为了考察分析系统和参数是否适合于研究体系。可以采用经论证和确认的替代化合物,建立和确认组成未知可沥滤物分析体系的每种分析技术进行系统适用性验证。系统适用性验证考虑的重点包括:
1)灵敏度,分析方法的定量限小于或等于AET。应对选择的替代参考物质分别评价,定量限的具体评价方法可参照已知可沥滤物指导原则,确保尽可能所有物质的检出限均小于AET;
2)特异性,即在样品中存在其他可预期成分的情况下,能够明确评估分析物的能力。评价方法对特定浓度一组替代参考物质进行分析,评价系统对这组物质的分离能力。
3)不确定因子(UF值),通常对GC/MS系统,可以直接采用2作为UF值。而对于LC/MS,由于浸提物不确定因子差异较大,通常考虑采用这一组参考物质单位浓度的响应值的差异(UF值)来评估AET。
未知可沥滤物分析体系的建立,往往采用一组经论述的替代参考物质,以验证分析体系的灵敏度、分离能力和不确定度。替代参考物质的选择可综合考虑以下因素:1)这些物质能够涵盖不同性质的化合物(包括极性、分子量大小等)。材料组成信息和加工工艺信息,可能会有益于替代参考物质的选择2)来自实验的官能团结构信息;3)保留时间的接近性;4)不确定因子等。适用时,还可选择商品化的混合标液作为替代参考物质的一部分。
对于元素分析体系,宜选择适当的元素混合物进行系统适用性评估,元素混合物的选择可以考虑所研究器械材料、工艺引入的元素,也可考虑参考ICH Q3D选择所关注元素。选择适宜的涵盖高中低质量数多个内标以尽可能确保其质量轴在全波段内响应校正的准确性。除此之外,对于Si, S,P 等特殊元素可能需要考虑特定方法进行准确表征。
在整个表征过程中,除了建立未知可沥滤物分析方法外,还应考虑一些具有较强毒性的特殊物质,如亚硝胺类化合物、多环芳烃类物质等,如果某些材料组成或工艺等信息表明存在有该类化合物的可能性时,可能需要针对这类物质开发专属的分析方法。因此该类物质毒性较强且往往是痕量水平,故对分析灵敏度要求较高,同时某些化合物结构本身对通用型检测器的响应较低。
2.方法学考察
对任何物质的分析方法均应考察其方法学的可靠性,未知可沥滤物分析同样不例外。不同于已知可沥滤物测定时对方法的验证(verification)和确认(validation)的要求,由于未知可沥滤物潜在分析物群体庞大且多样,因而单一方法不适用于所有潜在分析物,并且单一方法也无法获得所有潜在分析物的高度准确和精确的浓度估计值。因此,在可能的情况下,应采用一组能够代表本产品潜在分析物整个群体的替代分析物对用于筛选的分析方法进行界定(qualified),以确保分析方法适合于其预期用途。
未知可沥滤物分析方法的方法学考察参数不同于已知可沥滤物分析,一般包括重复性、线性、回收率等,具体可见ISO 10993.18-2020附录F的要求。
3. 定性及定量分析
对于超过AET的物质应进行定性和定量分析,未知可沥滤物定性过程是一个复杂的过程。尽管某些商业化的质谱库可以为未知可沥滤物的定性提供便利,但实验室建立未知可浸提物和可沥滤物数据库(E&L数据库)能更快更准确的获得未知可沥滤物的信息,附录B给出了数据库建立的一般思路供参考。即便在有数据库可供参考的前提下,定性过程也要更多的依赖于分析人员对于待测样品的识别及结构解析的能力,同时也需要几种分析手段互相印证,研究人员应保留整个定性推导的全过程,虽然详细推导过程可能不一定作为申报资料。
定量分析可分为半定量(semi-quantitative analysis) 、或定量(quantitative analysis),对于超过AET并完成定性分析的物质可选择半定量方法评估其释放量,半定量可根据方法学考察过程中选择的内标,也可以选择保留时间接近或者结构类似的参考物质进行半定量分析。如果采用准确定量分析,应参照《已知可沥滤物测定方法验证及确认技术审查指导原则》给出的方法学验证参数,对分析方法进行全面的验证。
在半定量研究时,建议在替代化合物的保留时间范围内,采用多个内标物进行半定量,可以考虑所研究器械中可能存在的浸提物作为内标。同时,确认每种分析技术的系统不确定因子UF(见ISO 10993.18-2020 附录E.3)。对于GC技术,UF可以考虑用2,对于LC技术,一般可采用混合参考物相对标准偏差(RSD,%)计算获得,即:UF=1/(1-RSD)。
当然,一个好的内标物一般应具有以下特点:1)与选择的分析技术/分析方法相兼容;2)在相应分析方法中有好的“行为表现”。例如,在GC分析中,内标物不应该有明显的拖尾,也不应发生不可逆的柱吸附;3)应在分析基质中稳定;4)不能干扰待测溶液中其他成分的分离和检测结果;5)应当与其他被测组分有相似的响应值。
(五)浸提物的表征
浸提物质的分析应考虑有机物和无机物。可根据挥发性将有机可浸提物定性分成三类;挥发性有机化合物(VOC)、半挥发性有机化合物(SVOC)和非挥发性有机化合物(NVOC)。
由于潜在浸提物种类的复杂性,单一分析手段并不能对所有可浸提物实现有效的定性和定量分析,需要采用多种分析技术完成未知可沥滤物的研究。常用的分析技术包括气相色谱、液相色谱、ICP。其中气相色谱和液相色谱及质谱主要用于分析有机浸提物,电感耦合等离子体原子发射光谱(ICP-AES)和电感耦合等离子体质谱(ICP-MS)主要用于分析元素可浸提物。
有机可浸提物分析取决于有机物的性质,没有一种色谱方法可适用于所有潜在有机可浸提物。例如顶空取样气相色谱(HS-GC)通常用于分析易挥发性有机物,液体进样气相色谱质谱(GC-MS)通常用于分析挥发性有机化合物(VOCs)和半挥发性有机化合物(SVOCs),液相色谱(LC)和液相色谱质谱(LC-MS)通常用于分析SVOCs和不挥发性有机物(NVOCs)。由于一种浸提液可含所有不同种类和浓度的化合物,因此需要采用上述技术的联合应用(例如,色谱法与多种检测器结合、NMR和其它光谱技术等),并充分考虑色谱技术、灵敏度等因素的影响。
ICP-AES和ICP-MS分析手段适用于浸提液中元素分析,而这种元素通常可以以有机和无机两种形态存在。ICP分析的一个潜在缺点是它不能揭示元素存在的形态,例如,硫可被浸提为元素硫,硫酸盐离子,或作为有机可浸提物的一部分(如巯基苯并噻唑),但是这三种形态物质的毒理学风险可能并不相同。但这并不意味着所有元素都需要进行形态研究。因此,需要结合收集到的材料组成及加工工艺信息和有机可浸提物分析结果,确认是否需要进一步实验研究。
某些情况下,如需要,离子色谱法(IC)可用于可浸提物筛选,以分析可浸提的无机阴离子(例如,氟化物、氯化物和硫化物)和低分子量有机酸(例如,醋酸和甲酸)。
上述分析技术除了用于不同性质的未知可沥滤物以外,不同分析技术获得结果还可以用于相互印证。
除了采用上述特异性分析方法进行表征外,往往还需要采用一些非特异性分析,例如:总有机碳(TOC)、傅立叶红外光谱扫描、pH。尽管这些分析方法并不对可浸提物进行定性和定量分析,但是这些分析手段往往可以评估某类物质总量,但是会有利于对特异性实验的结果进行辅助性信息输入,成为与特异性表征结果互相支持的证据。传统的化学分析项目,如还原物质、紫外吸光度、酸碱度、蒸发残渣、重金属可能在一定程度上给出非特异性可沥滤物信息,但对未知可沥滤物表征的贡献是有限的。
常见的表征分析方法见表X
|
材料类型 |
特性 |
方法举例 |
定性 |
定量 |
|
所有类型 |
有机可浸提物(VOC) |
HS-GC或GC与FID和/或MS |
X |
X |
|
总有机碳(TOC)a |
─ |
X |
||
|
有机可浸提物(SVOC) |
HS-GC和GC与FID和/或MS |
X |
X |
|
|
HPLC与UV、CAD、ELSD 和/或MS |
X |
X |
||
|
总有机碳(TOC)a |
─ |
X |
||
|
NMR |
X |
X |
||
|
有机可浸提物(NVOC) |
HPLC与UV、CAD、ELSD 和/或MS |
X |
X |
|
|
NMR |
X |
X |
||
|
总有机碳(TOC)a |
─ |
X |
||
|
非挥发性残留物 |
─ |
X |
||
|
元素可浸提物 |
ICP-AES,ICP-MS a |
X |
X |
|
|
阴离子与阳离子 |
离子色谱法 |
X |
X |
|
|
a通常使用水浸提溶剂(例如水、盐水)机玻璃块制成,具 |
||||
需要说明的是,为满足毒理学风险评估要求,在浸提研究早期与毒理学家的密切合作是必要的,以便于进一步确认提取物表征的充分性。例如,硅橡胶是医疗器械中常用的医用材料,并且在植入性医疗器械中应用广泛。硅橡胶在有机相溶剂进行浸提时,通常会发现硅氧烷类化合物,其中可能包括线性和环状硅氧烷。由于环状硅氧烷与线性硅氧烷较大的毒理学特性差异,在浸提液化学表征过程中应尤其引起注意,除确切表征其官能团结构外,宜对低分子环状硅氧烷进行准确定量或半定量。再比如,某些聚合物的可浸提物会产生较多的低聚物,根据毒理学家评估的结果,某些情况下,可以作为一组化合物进行半定量处理。此时就需要和毒理学家进行密切合作,以判断是否需要对浸提物进行更进一步的分析。
(六)可沥滤物研究(Leachable study)
1.需开展可沥滤物研究的情形
对于相当数量的医疗器械,如齿科医疗器械、骨科用医疗器械及其他很多植入性医疗器械,包括一些短期和长期(prolonged)接触的器械,根据可沥滤物的定义,一般无法通过实际的可沥滤物研究来评估接触量。大部分情况下依赖于浸提研究完成可沥滤物评估。基于浸提研究进行的临床释放量评估以确认其安全性,不存在潜在毒理学风险,则一般不必进行进一步的可沥滤物研究。即使是对于有可能通过临床实际来完成可沥滤物研究的器械,如某些分析背景较为简单的间接接触的器械,也仍然适用于上述原则。
然而,如果前述可浸提物研究结果不能支持毒理学评估,则有必要对医疗器械进行更实际的估计。这种更实际的估计通过模拟浸提条件或使用持续时间短于临床使用的加速浸提条件实施。浸提研究阶段需根据器械的特性及临床使用性质对浸出研究方案设计及参数选择进行系统论述,以证明所采用的浸提研究可以充分表征器械使用过程中的可沥滤物接触状况。
以下情况有可能需要进行可沥滤物研究:
1)浸提研究中发现某些浸出物基于临床释放量评估,存在潜在安全性危害时,则可考虑采用更接近实际接触方式的评估,采用临床实际、或采用临床实际接触介质模拟或加速浸提进行可沥滤物研究。值得注意的是采用临床实际接触介质模拟或加速浸提严格意义上来讲,仍然是浸提研究的内容,但由于绝大多数实际接触介质分析背景的复杂性,导致不能直接将浸提溶剂开展未知可沥滤物扫描分析。因而,通过上述采用替代溶剂法获得的浸出物谱,通过定性定量研究,存在潜在毒理学风险时,则可进入已知可沥滤物研究来进一步评估。该情况下可沥滤物研究详细内容见《已知可沥滤物测定方法验证及确认技术审查指导原则》。
2)某些情况下,适用时,如与某些药液接触的器械,如果预计所接触药物与器械可浸提物发生相互作用而可能产生浸出物谱以外的可沥滤物,则可考虑在可沥滤物研究时,通过扫描方式获得额外可沥滤物信息。由于基于实际接触介质进行扫描的复杂性和局限性,宜对所采用的实验方案,包括可能采用的替代溶剂或替代介质的转换方案进行充分阐述。除此之外,比较接触介质(如药液)与器械接触前后可沥滤物谱的变化也是提供有效信息的方式之一。
3)某些情况下,适用时,如某些可降解医疗器械,其降解产物或其他反应副产物如果预计在浸提研究中无法获得,当结合相关标准(如GB/T 16886.9,GB/T 16886.13等),在模拟液或实际接触介质中开展降解产物/反应产物研究时,考虑通过扫描方式获得额外可沥滤物信息。虽然,严格来讲,这部分研究也不是真正意义的可沥滤物研究。同样,基于所采用介质进行扫描的复杂性和局限性,宜对所采用的实验方案,包括可能采用的替代溶剂或替代介质的转换方案进行充分阐述。
4)某些情况下,适用时,尤其是器械临床实际接触介质较为简单时,如注射用水、生理盐水等,也可以考虑采用实际接触介质直接进行可沥滤物扫描,而不一定采用替代溶剂。
2.可沥滤物的风险评估
可沥滤物研究一般包括目标可沥滤物溶出量研究(详细研究指南见《已知可沥滤物测定方法验证及确认技术审查指导原则》)和通过扫描方式,即未知可沥滤物分析体系,对高于AET的可沥滤物的定性定量研究。
可沥滤物研究的结果将被用于支持总体的生物学评价(详细标准或指南见GB/T 10993.1和《医疗器械生物学评价策略及使用指南》、毒理学风险评估(详细标准或指南见GB/T 16886.17和《已知可沥滤物允许限量建立及风险判定技术审查导原则》、其它风险管理过程中的评估,如某些变更评估(详细标准或指南见YY/T 0316 和《医疗器械材料等同性指南》等。
3.采用的表征方法
已知可沥滤物研究的表征方法及技术详见《已知可沥滤物测定方法验证及确认技术审查指导原则》。
如需进行未知可沥滤物研究,如上述“可沥滤物研究的应用”中2)-4)的情景,则可采用的分析方法与第(三)章浸提研究时采用的方法基本类似。所不同的是未知可沥滤物的分析,由于实验样本的背景更加复杂,可能会大大降低分析系统的灵敏度及特异性等,因而,实验体系更加依赖于色谱、光谱及其质谱联用等技术。某些情况下,可能需要引入一些样品处理技术,如,可能涉及到的液液萃取技术及萃取验证等。
4.可沥滤物方法学研究
通过信息收集、文献研究、未知可沥滤物鉴别等手段获得的目标化合物信息,需要进行进一步定量研究的方法学要求参见《已知可沥滤物测定方法验证及确认技术审查指导原则》。
对于需要进行未知可沥滤物扫描的情形,方法学评估参见本指南“4 未知可沥滤物分析体系分的建立”
(七)未知可沥滤物体系应用于材料成分表征
在信息收集阶段或生物学评价的其他阶段,某些情况下,若需要通过实验获得构成器械的材料或组件成分定性定量信息时,采用溶剂溶解、消解、多溶剂极限浸提等方式的测定,可参照浸出研究相关章节进行。所不同的是以材料成分表征为目的的浸出研究,其溶剂的选择不一定依赖于临床接触介质的性质。
四.报告要求
· 信息描述: 应根据产品说明书,生产工艺资料、供应商提供的信息等材料,描述器械的结构及材料组成、拟研究材料/器械的理化特性及与可沥滤物相关的文献信息、必要的器械的生产工艺中加工助剂及灭菌信息、临床应用信息等。这些信息用于作为相容性研究的实验设计、参数设计的论述依据。
· 对浸提实验设计论述:包括规格的选择(如适用)、溶剂的选择、浸提参数(浸提时间、浸提方式、浸提比例等)的选择及确认
· 浸出物表征方法:包括任何对浸提液的前处理描述(如溶剂转换),所采用的仪器设备及其系统适用性(灵敏度、选择性、不确定因子等)的选择依据及结果(如典型图谱等)
· 定性分析过程及结果:分析评估阈值的确定,可浸出物的鉴定结果。适用时,提供可提取物的典型图谱。必要时,提供几种分析手段互相印证的论述。
· 定量分析及结果(包括半定量、定量)。对浸出研究结果进行总结和归纳,最好以表格的形式列出可浸出物清单。清单中可列出浸出物的化学名称、分子量、结构式或CAS号(如有)、定量结果、可能来源等信息。结构或官能团类似物可合并列出,便于毒理学风险评估。适用时,论述特殊关注物质的定量分析的方法学及结果。
· 根据临床使用情况,将浓度换算为器械的临床接触评估量(如,ug/器械)
五.关键性术语
1.扫描分析方法(analytical screening method):在未知可沥滤物扫描模式下对所有相关物质的发现、鉴别和半定量研究等的分析方法。
2.目标分析方法(analytical targeting method):采用经验证及确认的方法对已知分析物质进行准确定量研究的分析方法。
3.半定量分析(semi-quantitative analysis):通过一种或多种最接近标准物质的替代物的浓度响应分析拟研究物质浓度的一种分析方法。
备注:与半定量分析对应的是,部分文献资料中还有一种估计性定量分析(estimated quantitative analysis),严格意义上估计性定量不是一种定量研究的方式,只是对不同情况下半定量研究(不管是否考虑了其响应因子情况)的一种方法概述。
4.浸提物(extractable):在实验室浸提条件及参数下对医疗器械或材料采用浸提方式获得的物质。
备注:由于翻译差异,部分领域或部分标准等文献资料中将extractable翻译为“提取物”“释放物”“萃取物”等。
5.可沥滤物(leachable):医疗器械或材料在临床使用条件下释放出来的物质。
备注:由于翻译差异,部分领域或部分标准等文献资料将leachables翻译为“浸出物”“可析出物”“释放物”等。
六.参考文献
1.ISO10993-18:2020,Biological evaluation of medical devices —Part 18: Chemical characterization of medical device materials within a risk management process [S/OL].[2020.01]/[2020.05]
2.ISO10993-1:2018, Biological evaluation of medical devices —Part 1: Evaluation and testing within a risk management process [S/OL].[2018.]/[2020.05]
3.Dennis Jenke,Norman Liu.Chromatographic considerations in the standardization of liquid chromatographic methods used for extractables screening[J].Journal of liquid chromatography & related technologies,2016,39(13):613-619
4.Lei Tian, Lan Lin, Stéphane Bayen.Optimization of the post-acquisition data processing for the non-targeted screening of trace leachable residues from reusable plastic bottles by high performance liquid chromatography coupled to hybrid quadrupole time of flight mass spectrometry[J].Talanta,2019(193):70–76
5.Pauline Legrand , Anne Desdion, Gaétan Boccadifuoco,et al.Development of an HPLC/UV method for the evaluation of extractables and leachables in plastic: Application to a plastic-packaged calcium gluconate glucoheptonate solution[J].Journal of Pharmaceutical and Biomedical Analysis,2018(155):298-305
6.Dennis Jenke.Identification, analysis and safety assessment of leachables and extractables[J].Trends in Analytical Chemistry,2018(101):56-65
7.Dennis Jenke.Identifying and Mitigating Errors in Screening for Organic Extractables and Leachables: Part 3−−Considering Errors of Implementation and the Use of a Database to Judge and Promote Good Science and Efficient Practices[J].PDA Journal of Pharmaceutical Science and Technology,2020,74(1):134-146
8.Piet Christiaens, Jean-Marie Beusen, Philippe Verlinde, et al.Identifying and Mitigating Errors in Screening for Organic Extractables and Leachables: Part 1-Introduction to Errors in Chromatographic Screening for Organic Extractables and Leachables and Discussion of the Errors of Omission[J].PDA Journal of Pharmaceutical Science and Technology,2020,74(1)::90-107
9.Dennis Jenke, Piet Christiaens, Jean-Marie Beusen, et al. Identifying and Mitigating Errors in Screening for Organic Extractables and Leachables: Part 2-The Errors of Inexact Identification and Inaccurate uantitation[J]. PDA Journal of Pharmaceutical Science and Technology, 2020,74(1):108-133 .
10.Dennis Jenke.Identification and Quantitation Classifications for Extractables and Leachables[J].PDA Journal of Pharmaceutical Science and Technology,2020,74(2):275-285
11.Dennis Jenke,Alex Odufu.Utilization of Internal Standard Response Factors to Estimate the Concentration of Organic Compounds Leached from Pharmaceutical Packaging Systems and Application of Such Estimated Concentrations to Safety Assessment[J].2012(50):206-212
12.Steven A. Zdravkovic, Cindy T. Duong, Ashley A. Hellenbrand,et al.Establishment of a reference standard database for use in the qualitative and semi-quantitative analysis of pharmaceutical contact materials within an extractables survey by GC–MS[J].Journal of Pharmaceutical and Biomedical Analysis,2018(151):49-60
13.FDA. Use of International Standard ISO 10993-1, Biological evaluation of medical devices —Part 1: Evaluation and testing within a risk management process. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-international-standard-iso-10993-1-biological-evaluation-medical-devices-part-1-evaluation-and.[2016.06/2020.05].
14.马玉楠,杨会英,高用华等译.浸出物和可提取物手册-吸入制剂的安全性评价、界定与最佳实践[M].北京:化学工业出版社,2014.
15.中国医药包装协会.药品与包装相容性理论与实践[M].北京:化学工业出版社,2019.
附录A
未知可沥滤物研究的一般程序
附录B
未知可沥滤物研究的数据库建立一般思路
一、概述
医疗产品中未知可浸提物和可沥滤物(extractables and leachables, E&L)的研究通常从采用色谱-质谱联用的技术对样品中未知可沥滤物谱进行全谱扫描开始[1-2],并对扫描的未知可沥滤物进行定性定量分析。整个未知可沥滤物分析过程面临诸多挑战,包括:初步的未知可沥滤物谱扫描是否发生遗漏,鉴别是否正确,定量分析是否准确等。由于E&L化合物的特殊性以及彼此结构的关联性,通过建立专门数据库的方法来应对上述挑战成为可能,并且近年来渐渐被医药行业的相关研究者逐步推广。
二、建立可浸提物和可沥滤物专门数据库的必要性
对未知可沥滤物解析需要通过对已有知识的检索来实现,也就是依赖数据库。在医疗产品未知可沥滤物E&L的初期,一般会利用普适性的数据库:如,标准或商业化软件质谱数据库,由于质谱是未知可浸提物和可沥滤物研究的最主要检测手段,通过对通用质谱库的检索和比对可以快速锁定可能的化合物结构。使用庞大的普适性数据库鉴别未知可沥滤物,通常是通过比较未知可沥滤物和库中化合物谱图的匹配度(%),从而给出匹配度最高的化合物。
由于数据库比较庞大,而使用的分析方法有一定差异,尤其是没有其他分析化学信息辅助印证的情况下,有时候很容易出现误判。图1对比了使用商业软件数据库和实验室内部建立的专门的可浸提物和可沥滤物数据库(下文统称:E&L数据库),对同一个未知可沥滤物质谱图匹配的结果[3]。在这个例子中采用内部E&L数据库解析并验证了未知可沥滤物结构的正确性。从图1的比较可以看到,E&L内部数据库和商业数据库相比,内部数据库给出了化合物的准确结构、较高的匹配分数,还包含了“保留时间”这个额外的匹配印证信息,使得未知可沥滤物鉴别具有更高的可靠性。相反,仅仅依赖于普适库鉴别获得的结果,与内部数据库比较,得到了完全不同的另一个化合物。由此可见,针对可浸提物和可沥滤物建立专门的数据库对于准确、高效、全面地研究未知可沥滤物和未知体系十分必要。
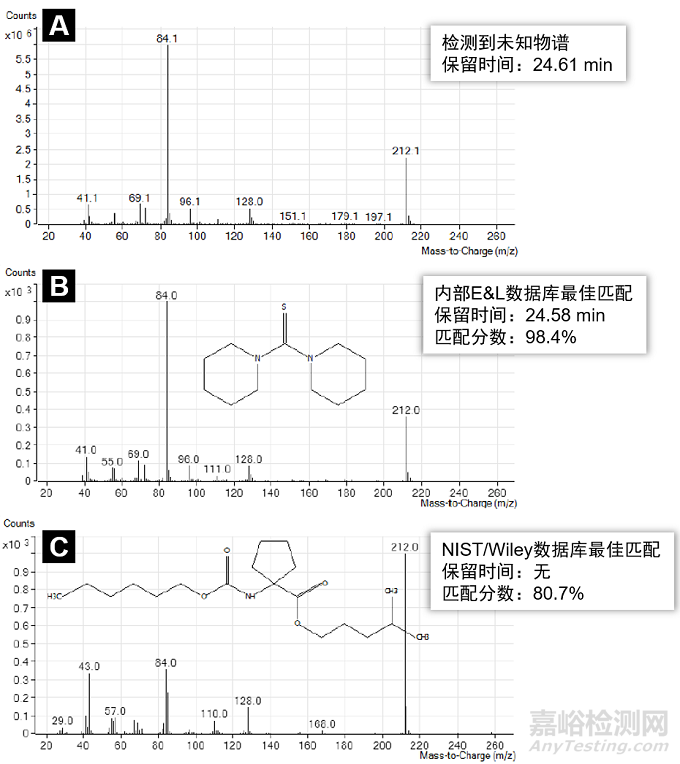
图1 [3]. (A) 未知可沥滤物质谱图(电子电离EI),GC/MS 中该物质的保留时间为24.61 min。 (B)通过检索实验室内部E&L数据库得到的正确结构,匹配分数和保留时间都非常接近100%匹配。
(C)通过检索商业软件NIST/Wiley数据库EI图谱的错误结果,没有保留时间信息作为参考,最佳匹配分数80.7%只能判定为 “不准确的鉴别” 。
三、E&L数据库建立的一般思路
Jenke,D. 等人在对未知可浸提物和可沥滤物分析的综述中将E&L数据库定义为:一个对于涵盖了实验室测试中遇到过的以及预期可能遇到的可浸提物和可沥滤物的必要信息的集合。这里“必要信息”至少包括可以准确鉴别这些化合物并进行准确定量的分析化学信息,例如代表性质谱图谱及指纹信息,色谱的保留时间、响应因子等[4]。
实验室建立E&L化合物及材料数据库一般是从少数几种熟悉的材料,几十种化合物开始的。
建立和评价一个数据库的可以从以下四方面考虑:数据库中化合物数目及与所研究器械的相关性,E&L信息的种类和数目,数据库中信息的可信度[7]、数据库的补充及完善。
(一)数据库中化合物数目与所研究器械的相关性
考虑到数据库的主要目的是分析未知的可浸提物/可沥滤物,所以简单来看,数据库中所收录的化合物越多,遗漏就越少。但单纯的用数目衡量是片面的,更需要关注的是数据库中的化合物在多大程度上涵盖了最常见、关键的可浸出物/可沥滤物。尤其是本实验室经常涉及到的样品体系的可浸出物/可沥滤物,例如某一类医疗器械或某一类高分子材料的药包材[4] 。一个有效的E&L数据库通过容纳大量的与研究体系高度相关的化合物以及相应的分析方法可以将未知可沥滤物的扫描(screening)过程在某种意义上变成目标物分析(targeting)的过程[9]。同时由于数据库的专业性和全面性而更大程度上减少了遗漏和错判,如下图2所示。通过可靠的未知可沥滤物分析体系,结合可靠的未知可沥滤物数据库,可以降低可浸出物谱鉴别结果的不确定。
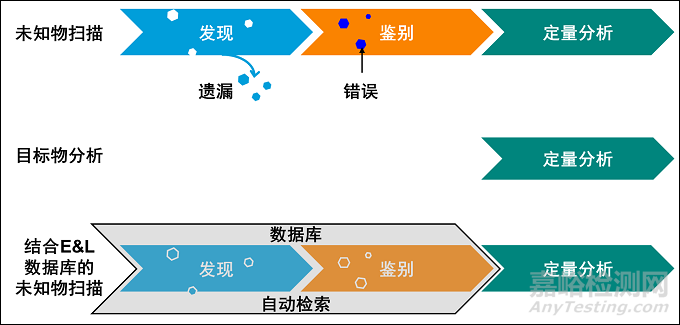
图2. 未知可沥滤物扫描和目标化合物分析。
另外值得一提的是,数据库是动态的,随着实验室分析的的器械、材料的种类的积累,不仅数据库中E&L化合物数目、种类会增加,方法也会相应完善[5]。因而这也是行业内力求将数据库进行的统一化和标准化的难点。由于每个实验室所评价的器械的不同,各自实验室可能会根据实验室器械材料、工艺等特点,首先会建立满足本实验室评价要求的数据库。
(二)E&L信息种类、数目及其应用
对于数据库中每一个化合物的结构和名称、所用的分析方法及该化合物对应的特征性响应参数(如色谱的保留时间)、谱图是最基本的信息。在此基础上加入越多的如CAS号、相对响应因子(RRF,尤其对于液相色谱方法)、对化合物的多个互补印证分析方法等,任何有利于在未知可沥滤物扫描过程中更精准地鉴别未知可沥滤物的信息,都能使数据库更加完善、有效。
由于产品的E&L表征,是医疗器械生物学评价的一部分,最终目标是服务于毒理学风险评估。因而,当实验室在实践当中,对E&L数据库建立并积累了足够的经验,其化合物的毒理信息也将成E&L数据库的重要组成部分。E&L数据库纳入化合物的毒理信息,对未知可沥滤物的鉴别和定量过程,也就是未知可沥滤物的表征程度,起到非常重要的作用。Jenke D. 和Carlson T. 在参考文献[6] 中强调了未知可沥滤物安全性评估在早期,未知可沥滤物E&L研究过程中可以通过免去对毒性小而在谱图中有响应物质的鉴别和定量,从而提高未知可沥滤物分析效率。
表1 [6].包含毒理学信息的E&L物质数据库示例
|
化合物名称 |
CAS号 |
毒理信息a) |
毒理学不确定因子c) |
风险系数mg/天d) |
||||||
|
耐受量mg/kg b) |
类别e) |
途径 |
动物 模型 |
T1 物种间 |
T2 物种内 |
T3 途径 |
T4 其他 |
|||
|
Octyldecyl phthalate |
119-07-3 |
44070 |
LD50 |
口服 |
大鼠 |
10 |
10 |
10 |
10 |
308 |
|
Dibutyl phthalate |
84-74-2 |
720 |
LD50 |
静脉 |
小鼠 |
10 |
10 |
1 |
10 |
50.4 |
|
Cyclohexanone |
108-94-1 |
100 |
NOEL |
静脉 |
大鼠 |
10 |
10 |
1 |
1 |
70.0 |
|
Dodecanol |
112-53-8 |
390 |
LD50 |
静脉 |
大鼠 |
10 |
10 |
1 |
10 |
27.3 |
|
8000 |
TDLO |
皮下 注射 |
豚鼠 |
10 |
10 |
10 |
10 |
56.0 |
||
|
100 |
NOAEL |
口服 |
大鼠 |
10 |
10 |
10 |
1 |
7.00 |
||
a) 原文数据库中还列出了每一个化合物毒理信息的出处参考资料,此处为简化表格略去了。
b)可允许的每日耐受量。
c)每个类别(T1 – T4)的不确定性因子范围为1-10。
d)风险系数在原文中定义为表中第三列每日毒理学的耐受量乘以70 kg体重再除以四个不确定性因子(T1 – T4)的乘积。
e)LD50:半数致死量,NOEL:无明显作用剂量,TDLO:最低(有报道的)中毒剂量,NOAEL:无明显不良作用剂量。
f)原文数据库中也包含了每个物质的致癌性和致突变性信息作为参考,但未进行深入讨论,此处为简化表格略去了。
另一个方面是数据库中包含的分析方法种类的多样性。此前已经提到了E&L数据库不仅要总结化合物,更要包含用于识别化合物的分析方法。由于未知可沥滤物研究的复杂性,仅依赖于一个方法一般不太可行,会增加了遗漏和错判的风险。例如参考文献5列举了包含同一化合物在多种分析方法下相对响应因子(RRF)的数据库示例(参见表2及表3)。以表中苯乙酮为例,虽然利用三种互补的分析方法均可以识别出样品中苯乙酮的存在,但是HS-GC-MS和LC-MS中苯乙酮的响应非常弱[3]。这意味着:首先只有当苯乙酮含量较高时才能利用这两种方法看到该可沥滤物的存在,也就是说,当苯乙酮浓度较低时,如果所选择的分析方法为上述两种方法时,很容易将这个化合物遗漏,这也是为什么在未知可沥滤物扫描体系中要采用多种互补的分析手段的原因;其次,采用HS-GC-MS或者LC-MS对苯乙酮定量分析的误差会很大(由于响应不灵敏),因而,定量、半定量分析均应采用GC-MS的方法[3]。
表2 [6].数据库中对比不同互补分析方法下同一种化合物RRF示例
|
CAS号 |
化合物名称 |
不同分析方法中的相对响应因子(RRFs)a) |
||
|
HS-GC-MS |
GC-MS |
LC-MS (APCI) |
||
|
95-16-9 |
Benzothiazole 苯并噻唑 |
0.003 |
0.901 |
1.209 |
|
100-52-7 |
Benzaldehyde 苯甲醛 |
0.042 |
0.699 |
0.178 |
|
98-86-2 |
Acetophenone 苯乙酮 |
0.033 |
0.690 |
0.009 |
|
761-65-9 |
N,N-Dibutylformamide N,N-二丁基甲酰胺 |
0.001 |
0.559 |
1.206 |
|
1119-40-0 |
Pentanedioic acid, dimethyl ester 戊二酸二甲酯 |
0.001 |
0.460 |
0.245 |
a) 加粗的数值对应的分析方法可以给出对应化合物最准确的定量分析信息。
表3 [3]. 数据库中对比不同互补分析方法下同一种化合物RRF示例
|
CAS号 |
化合物名称 |
不同分析方法中的相对响应因子(RRFs)a) |
||
|
HS-GC-MS |
GC-MS |
LC-MS (APCI) |
||
|
95-16-9 |
Benzothiazole 苯并噻唑 |
0.003 |
0.901 |
1.209 |
|
100-52-7 |
Benzaldehyde 苯甲醛 |
0.042 |
0.699 |
0.178 |
|
98-86-2 |
Acetophenone 苯乙酮 |
0.033 |
0.690 |
0.009 |
|
761-65-9 |
N,N-Dibutylformamide N,N-二丁基甲酰胺 |
0.001 |
0.559 |
1.206 |
|
1119-40-0 |
Pentanedioic acid, dimethyl ester 戊二酸二甲酯 |
0.001 |
0.460 |
0.245 |
a) 加粗的数值对应的分析方法可以给出对应化合物最准确的定量分析信息。
(三)数据库中信息可信度
利用数据库来研究未知可沥滤物的前提是数据库的信息要准确并严谨可信,而“准确“主要体现在化合物鉴别上。由于数据库的结构错误或者结构和相关化学信息错误对应,造成的未知可沥滤物错判会直接导致误导后续毒理风险评估。因此,在建立数据库的时候通常需要“三重印证”或至少“两重互补印证”(orthogonal methods)[6]。具体而言,就是数据库中化合物要通过两种互补的分析方法互相印证,确定为同一种化合物,这是一种“两重互补印证”。而数据库中化合物经过了和参考标准品(reference standard)对比确认的过程。可以称为“三重印证” [3]。虽然在利用数据库对未知可浸提物/可沥滤物进行鉴别时,最理想的情况是三重印证,但是由于未知可沥滤物分析实际情况的挑战性,例如某些情况下,无法通过多种分析方法印证,或者无法找到参考标准品,可能采用其他基于图谱解析、信息收集、文献研究等的论述,也是一种有益的相互印证手段。
(四)E&L数据库的补充及完善
未知可沥滤物研究,未知可沥滤物的遗漏是潜在风险的最主要来源。因而,在数据库的基本结构和框架搭建起来后,可以考虑进一步对数据库进行内部归类和结构调整,在实践中对其进行优化和完善。例如,加入内部子结构的数据库,从而形成一个更加完善的数据库。这类子结构数据库包括:
1.同类材料的关联性数据库:
文献[4]中引用了Irganox类抗氧化剂的例子。这类抗氧化剂常用在塑料、橡胶塞中,关于Irganox相关的降解产物作为可提取物研究和报道过[5]。如果将E&L数据库中和Irganox相关的化合物关联起来,在分析新的使用了Irganox抗氧化剂的产品或材料时,这个关联的E&L数据库就可以辅助判断。特别注意的是关联物质和所使用的提取溶剂有关,比如同一种材料,用水以及用乙醇/水来浸提,可能看到的化合物数目不同。这一点在建立关联性数据库的时候应予以考虑。
2.特殊物质数据库/特殊方法数据库:
这部分数据库包括了ISO /TS 21726 Biological evaluation of medical devices — Application of the threshold of toxicological concern (TTC) for assessing biocompatibility of medical device constituents中描述的特殊物质,由于具有较强的毒性,当需要进行相关研究时,则通常需要建立针对这些物质的高灵敏度的特殊方法。
另一种情况是,随着实验室在E&L分析方面经验和数据库的积累,对一些实验室常见材料体系可能含有的E&L物质或者对于常用的E&L未知可沥滤物研究体系的“漏洞”会有较为准确的把握。例如, Zweiben[12]在分析药包材胶塞对应的可沥滤物时,看到电子电离法得到最高m/z为103的一个峰,在标准的NIST谱库中搜索不到相应化合物,但是实验室可浸提物专门数据库中找到了完全一样的谱图,虽然这个谱图之前也没有被识别出对应结构。基于这样的信息,Zweiben进一步用不同的方法,包括用氨气作为碰撞气最终解出了结构。这一案例表明,未知可沥滤物鉴别不可过分依赖商品化的谱库,实验室根据经验、研究历史,建立起的专门数据库在E&L的分析中对于找到化合物可能更加有效。在Zweiben的上述研究中,实验室对产品标签所使用的油墨产生的E&L物质分析案例中,根据经验关注到了LC-MS (ESI电离法)未知可沥滤物扫描图谱中一个疑似二苯基甲酮的小峰。由于在通过紫外光固化的标签中二苯基甲酮为常用的光引发剂,该物质有很强的紫外吸收,但是其ESI中却不易电离,导致其在LC-MS中灵敏度较低。正是凭着经验,Zweiben发现了以“不含二苯基甲酮”来注册的产品打印标签中微量二苯基甲酮沥滤物[6] 。这个例子也提示我们,在建立通用型数据库的同时,对于某些确认通用数据库无法涵盖、共流出或灵敏度极低导致容易“漏掉”的化学物质,可以考虑纳入特殊物质/特殊方法数据库。这类数据库同样需要在实践中不断完善。
参考文献
1. Jenke, D. Identification, analysis and safety assessment of leachables and extractables. TRAC-Trend Anal. Chem. 2018,101, 56 – 65.
2. Dorival-García, N.; Carillo, S.; Ta, C.; Roberts, D.; Comstock, K.; Lofthouse, S.; Ciceri, E.; D’Silva, K.; Kierans, G.; Kaisermayer, C.; Lindeberg, A.; Bones, J. Large-Scale Assessment of Extractables and Leachables in Single-Use Bags for Biomanufacturing. Anal. Chem. 2018, 90, 9006 – 9015.
3. Christiaens, P.; Beusen, J-M.; Verlinde, P.; Baeten, J.; Jenke, D. Identifying and Mitigating Errors in Screening for Organic Extractables and Leachables: Part 2: The Errors of Inexact Identification and Inaccurate Quantitation. PDA J Pharm. Sci. and Tech. 2020, 74, 108-133.
4. Christiaens, P.; Beusen, J-M.; Verlinde, P.; Baeten, J.; Jenke, D. Identifying and Mitigating Errors in Screening for Organic Extractables and Leachables: Part 1—Introduction to Errors in Chromatographic Screening for Organic Extractables and Leachables and Discussion of the Errors of Omission. PDA J Pharm. Sci. and Tech. 2020, 74, 90-107.
5. Ball, D.J.; Beck, S.; Feilden, A.; Zhang, L.; Shaw, A.; Nagao. L.M. A Comprehensive Database Optimized for the Selection of Materials and Risk Assessment of Leachables and Extractables. Poster presentation.
6. Jenke. D. Identifying and Mitigating Errors in Screening for Organic Extractables and Leachables: Part 3—Considering Errors of Implementation and the Use of a Database to Judge and Promote Good Science and Efficient Practices. PDA J Pharm. Sci. and Tech. 2020, 74 134-146.

来源:中国器审


