您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-12-03 12:52
药物的质量研究与质量标准的制定是药物研发过程的重要研究内容之一,贯穿于研发的整个生命周期。在药物质量研究工作中,分析方法学的开发及验证是其重要的组成部分之一。分析方法开发验证的目的是判断所采用的分析研究方法是否科学、合理,能否有效控制药品的内在质量特性,做到质量可控。本文旨在和大家一起交流溶出度方法学验证内容的一般研究思路,如有存在表述不当之处还请各位批评指正。
溶出度方法学验证的步骤主要有:1)初步确定分析方法,UV法或HPLC法;2)制定验证的方案,包括前期文献材料调研、验证目的、验证项目及不同项目验证的可接受标准;3)开始验证工作,积累收集数据及相应图谱;4)对验证的结果进行判断,评价分析方法是否通过验证。
溶出度方法学验证的项目与其他分析方法基本一致,常规验证项目包括:专属性、线性及范围、准确度、精密度和耐用性等,方法验证的指导原则可参考中国药典、ICH Q2(A/B)、USP通则<1225>、<1226>、<1092>等。

1. 专属性
专属性系指在其他成分(如杂质、降解产物、空白辅料等)存在时,采用的分析方法能正确测定出被测物的能力。专属性测定环节,应分别分析加有杂质、降解产物等控制成分的样品和实际样品,比较两组测试结果,结果合格的标准应该为:空白溶剂对主峰的检测无干扰,不超过1%;主成分与有关物质完全分离,分离度r≥1.5;峰纯度符合相应规定。
辅料对专属性的干扰:空白辅料是指除了活性成分以外的所有辅料和包衣材料,还包括油墨和胶囊壳。具体操作方法可按处方比例配制空白辅料(含油墨或胶囊壳)的混合样品,将该混合样品溶解或分散在溶出介质中,然后向溶液中加入一定量药物,作为供试品溶液,可接受标准为:辅料(包括胶囊壳等基质)对主峰的检测无干扰,不能超过2.0%。
对于溶出实验方法而言,还需要特别注意的一点是:取样时所采用的过滤装置,如滤膜、滤头等,必须要经过药物的吸附验证,防止对测定结果产生一定干扰,这一部分应在溶出方法开发阶段做充分论证研究。
2. 线性和范围
可取对照品适量,按照标准方法配置一系列浓度的溶液。一般操作是在容量瓶中配成一定浓度的储备液,分别精密移取储备液适量,稀释成系列浓度的溶液,通常至少使用5个浓度点(参见<1225>),1225中说明:对原料或成品药(制剂)的含量测定:一般应在测试浓度的80-120%,该范围是应考虑的最小规定范围,若超出此范围,应有正当理由,主要是根据剂型的特点;对于溶出度试验,应为规定范围的±20%,例如如果是控释制剂,规定1h后达到20%,24h达到90%,它的验证范围应为标示量的0-110%。另外,若线性贮备溶液制备过程中为了增加药物的溶解度,可能会用到有机溶剂,除非经过验证外,有机溶剂的量均不得超过总体积的5%(v/v)。
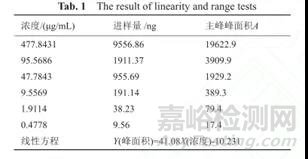
例如取头孢克肟对照品55.37mg,置100ml容量瓶中配置为储备液,然后就依次精密移取稀释成一系列梯度浓度,以浓度为纵坐标,相应峰面积为横坐标进行线性回归,结果表明头孢克肟浓度在0.48-477.84μg/ml范围内,进样量在9.34-9337.66ng范围内,进样量与峰面积呈良好线性关系。
3. 准确度
准确度即回收率实验。回收率试验目的是考察采用拟定方法测定结果与真实值或参考值接近的程度,且应应在规定的线性范围内进行试验。在回收率实验进行之前,USP1092建议:在回收率实验之前,过滤器、滤膜等对药物的吸附要进行全面评估,同时要设法排除由于仪器的玻璃材质部分对样品吸附而对测定结果造成的干扰影响。
具体的实验方法包括:在规定范围内,取同一浓度(相当于100%浓度水平)的供试品,用至少6份样品的测定结果进行评价;或考虑设计至少三种不同浓度,每种浓度至少平行配制3份,用至少9份样品的测定结果进行评价,回收率验证的浓度范围一般要求为限度的±20%。两种分析方法的选定应考虑分析的目的和样品的浓度范围。回收率供试样品溶液配制:按处方比例混合的空白辅料+不同浓度的主成分对照品或原料,再按照拟定的质量标准配制溶液,必要时可超声使主成分溶解。配制溶剂尽量与溶出介质体系一致。如果药物溶解性较差,可以将药物溶解在少量有机溶剂(一般不超过5%)中制备储备液,并用溶出介质稀释到最终浓度。可接受标准一般为:各浓度下的平均回收率应在98%-102%之间,相对标准偏差RSD应不大于2.0%。例如取头孢克肟对照品适量各三份,按照100%比例加入空白辅料,加溶出介质振摇溶解,作为50%、75%和100%供试溶液,回收率结果表明其方法回收率良好。

4. 重复性
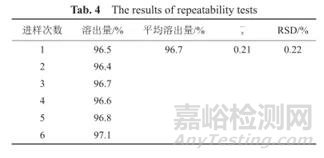
重复性即在同样的操作条件下,在较短时间间隔内,由同一分析人员测定所得结果的精密度。可在规定浓度范围内,取同一浓度(分析方法拟定的样品测定浓度,相当于100%浓度水平)的供试品,用至少6份样品溶液的测定结果进行评价;或设计至少三种不同浓度,每种浓度分别制备至少三份供试品溶液进行测定,用至少9份样品的测定结果进行评价(浓度设定应考虑样品的浓度范围)。实际实验操作中,可能有几种方法,方法一:取6个单独制剂分别测定溶出度,计算RSD,但该方法测定时受制剂个体差异影响比较大,如果测定结果重复性不好,可能是因为制剂含量差异所导致,用该方法时最好是挑选质量较好,例如含量均匀度较好的片剂进行实验;方法二即取供试品1片(粒),置于一个溶出杯中,按照溶出度方法测定,至规定取样点时去处六份供试液分别测定溶出度计算RSD值。结果接受标准为RSD不超过2.0%。例如取头孢克肟颗粒6袋,按照溶出度方法进行溶出,30min取溶出液滤过,进样计算溶出度,结果表明该溶出测定方法重复性良好。
5. 中间精密度
中间精密度即在同一实验室内的条件改变,如不同时间、不同分析人员、不同设备等测定结果之间的精密度。研究过程中的典型的变化,包括不同天、不同操作人员和设备。USP 1092中建议:可选用同一批次质量特征较好的制剂(如较好的含量均匀度)的溶出试验可以由同一实验室至少两个不同的分析人员进行,每个分析人员制备标准溶液和溶出介质和依据明确的提取和定量步骤进行。通常情况下,分析人员用不同的溶出液、分光光度计或HPLC(包括色谱柱)和自动进样器,在不同天进行试验。可接受标准:USP 1092建议:当该时间点的溶出量小于85%时,两个分析员溶出结果的平均值相差不得超过10%;当该时间点的溶出量大于85%时,两个分析员溶出结果的平均值相差不得超过5%。当然,具体的可接受标准可根据特定产品做具体规定。
6. 溶液稳定性
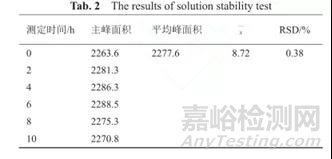
溶液稳定性考察的具体时间区间可根据不同的项目需求去做不同的考察。稳定性包括对照品溶液稳定性和供试品溶液稳定性。对照品溶液稳定性:取对照品溶液适量,在室温下放置,分别于不同时间点测定吸光度值,计算其RSD值;供试液稳定性:取自制样品适量,用相应介质制备成供试液,在室温下放置,分别于不同设置时间点测定吸光度值,计算其RSD值。对于UV法测定的供试液,一般稳定性做到24小时即可,缓控释制剂可相对延长时间;对于HPLC法测定的供试液,一般需满足一条溶出曲线所有样品测定完全的时间。如果溶液不稳定,还需要考虑温度(需要冷藏)、避光(透明容量瓶+棕色容量瓶)、以及容器材料(塑料或玻璃)等对稳定性结果的影响。可接受标准一般为:取每时间点的吸光度值,计算其RSD,应不大于2%,则说明该溶液在此时间段内的稳定性良好。
7. 耐用性
耐用性主要评估溶出条件故意做微小改变时对溶出方法耐用性的影响。对于该实验,最好选用具有较好质量特征(如具有较好含量均匀度)的制剂批次进行,排除制剂个体差异对该结果造成的干扰。HPLC法可根据具体情况考虑流动相组分差异、流速、PH值、色谱柱类型、分离温度、波长等变化对测定结果耐用性的影响;UV测定方法可结合不同项目溶出度方法的具体情况对表面活性剂浓度、pH值、溶出介质是否脱气处理、转速、温度、体积、取样时间、不同型号品牌的溶出仪等进行方法的耐用性研究,对比溶出条件的微小变化对产品测定结果的影响。例如若选择的溶出介质是缓冲液介质体系或是含有表面活性剂的介质体系,需要做pH值变化、表面活性剂浓度变化对溶出速度的影响,以确定溶出介质的耐用性。根据品种特点考察耐用性,推荐但不仅限于上述变动条件。
8. 溶出均一性
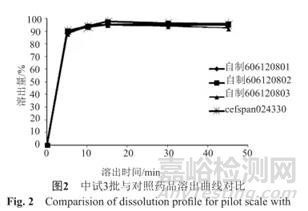
溶出均一性试验包括批内均一性和批间均一性。这两项指标既能检验药品本身质量特性是否符合规定,同时也可以检验溶出方法是否满足准确性、精确性良好的要求。批内均一性可取同一批次产品的6或12个剂量单位测定溶出曲线,计算各取样时间点的RSD值。其中,早期的一些取样时间点(如5min),要求RSD≤20%;其他时间点,要求RSD≤10%。批间均一性:取不同批次产品的6或12个剂量单位测定溶出曲线,比较各批次的溶出曲线是否相似。
综上,溶出方法验证的一般项目基本如上几项,当然并不局限于该些项目,具体的验证项目及可接受标准可根据产品自身特点所设定。
参考文献:
[1]. 《中国药典》2020年版四部<9101>:分析方法验证指导原则
[2]. USP通则< 1092>、< 1225>
[3]. 山广志,药物制剂质量研究——方法选择与验证
[4]. 胡利敏,杨丽,头孢克肟颗粒溶出曲线方法学验证[J]. 中国抗生素杂志,2017,5(42):373-376.

来源:药事纵横


