您当前的位置:检测资讯 > 行业研究
嘉峪检测网 2022-09-21 12:27
01印度医疗器械市场环境
近日,据印度医疗器械工业协会(AIM)公布的数据显示,2021年,印度医疗器械进口额从2020年的447.08亿卢比(约37.9亿元人民币)增长了41%,达到创纪录的632亿卢比(约53.6亿元人民币)。AIM一位工作人员表示:“这是一个令人担忧的情况,因为在六年的时间里,这一数字增长了五倍。”他说,2016年,印度进口了价值128.66亿卢比(约10.9亿元人民币)的医疗器械。
AIM的数据显示,中国仍是印度医疗器械的主要来源地,与2020年相比,2021年来自中国的医疗器械发货量增长了48%,达到135.38亿卢比(约11.48亿元人民币)。印度医疗器械市场估计超过877.52亿卢比(约77.4亿元人民币),其中海外供应商占70%以上。有专家表示,印度对进口医疗器械的依赖引发了许多担忧,许多医院寻求最新的医疗器械,这部分往往只能依赖进口。
今年以来,全球头部医疗器械厂家在印度动作频频:
今年3月,GE医疗印度宣布准备投资10亿卢比(1280万美元),建造位于印度的第四个制造工厂。
随后4月,西门子医疗印度承诺投资9.19亿卢比(1176万美元),用于制造CT和MR。
紧接着飞利浦也相继宣布收购10英亩土地以进一步投资印度。
跨国企业是全球价值洼地的最佳“狩猎者”,寻求利益最大化的区域进行投产经营,一直是他们赖以生存、维持竞争力的关键技能。因此,能够让他们相继押注、票选出来的地方一定存在独特的价值。
呼声之下,印度能否成为全球医疗器械产业链转移的下一站地?对中国医疗企业有哪些挑战与机遇?这些问题值得引起关注和注意。
想打入印度医疗器械市场,对其准入条件的把握成为先决条件。
02印度医疗器械的监管机构
印度卫生和家庭福利部(ministry of health & fw)下的中央药品标准控制局(central drug standards control organization,简称cdsco)为印度医疗器械的中央主管机构,其内部的重要组成机构印度药品管理总局(the drug controller general of india,简称dcgi)作为印度全国医疗器械政策的制定单位。
03印度医疗器械准入条件
(一)医疗器械的分类
2017年印度对医疗器械法规进行了大幅修订,公布了《医疗器械管理条例2017》(medical device rules 2017),从2018年1月起实施,参照了“全球医疗器械法规调和会”(the global harmonization task force,简称ghtf),将医疗器械分为A到D四类,对应从低风险到高风险四种风险级别。
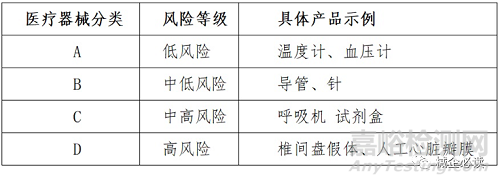
(二)医疗器械上市路径
MDR2017及其修正案将目前的医疗器械上市路径分为两种:公告型和非公告型。
公告型医疗器械:法规列出了37个具体的种类,强制要求制造商和进口商分别申请生产许可证和进口许可证。
非公告型医疗器械:除法规列出的37种类别之外的医疗器械。对于此类医疗器械,印度当局决定分步完成印度市场准入,分别为强制注册和生产或进口许可证申请。具体过渡阶段为:
A类和B类:2021年9月30日至2022年9月30日,要求强制注册,但是不强制申请生产许可和进口许可。
2022年10月1日起,强制要求生产许可&进口许可;
C类和D类:2021年9月30日至2023年9月30日,要求强制注册,但是不强制申请生产许可和进口许可;
2023年10月1日起,强制要求申请生产许可&进口许可;
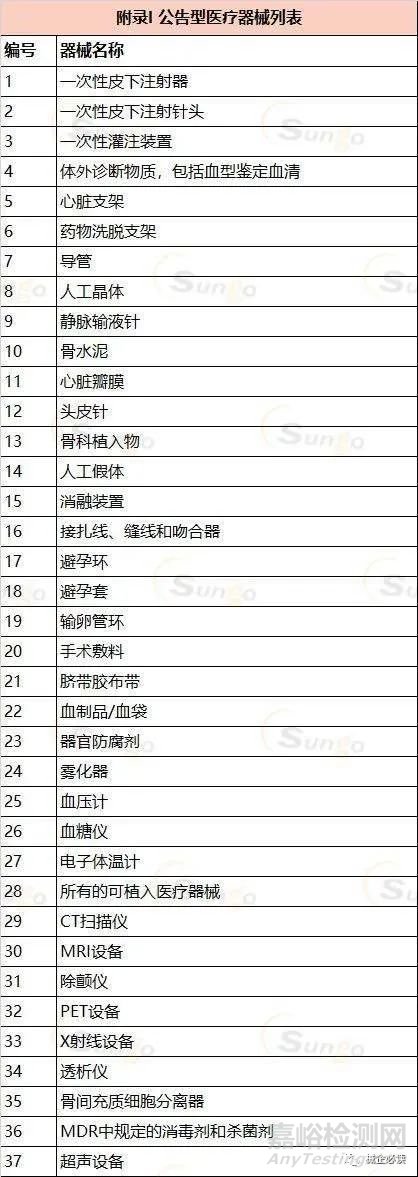
(三)生产许可申请
作为海外制造商,若想要将产品出口至印度,需要进行如下操作:
第一步:确定产品类别;
第二步:委派印度授权代表;
第三步:确保生产场地满足质量管理体系的要求;
第四步:准备满足MDR2017法规的申请文件;
第五步:印度官网提交申请文件;
第六步:在确认文件满足要求后,印度当局颁发生产许可证。
根据产品的分类,印度主管当局可能会在许可证颁发前后进行生产场地的审核。
(四)进口许可申请
出口医疗器械至印度进行销售或使用,必须经印度当地授权代理人(authorised agent)向cdsco提出医疗器械进口许可证申请(包括a至d级)。cdsco对医疗器械进口许可证的审核主要分为两步:
第一步:确认生产企业质量管理体系(quality management system,简称qms)的符合性
首先cdsco审核授权代理人所提交的生产企业质量管理体系,其必须符合印度医疗器械质量管理标准icmed 13485(修改采用自iso 13485,我国标准yy/t 0287等效采用了iso 13485),包括:生产企业qms技术文件、检测报告、最近一次现场检查报告等,如果cdsco认定生产企业qms的符合存在疑虑时,可以要求实施评估、产品检测或生产企业现场检查(费用由代理人承担)。
第二步:审核医疗器械的安全性与有效性
如果出口印度的医疗器械分类属于a级或者b级,授权代理人应向cdsco提交中国的自由销售证明,或者中国实施临床测试的数据(或其他可供证明产品安全性与有效性的数据)。如果出口印度的医疗器械分类属于c级或者d级,授权代理人必须在印度实施临床测试。
例外情况:如果医疗器械(A级至D级)已由欧盟、美国、加拿大、日本或澳大利亚的监管机构颁发了自由销售证明,则无需再进行临床测试。
(五)医疗器械标签
印度《医疗器械管理条例2017》第44条至48条对医疗器械的标签进行了要求。
医疗器械标签要求:该条例第44条规定,应用不可抹掉的墨水在医疗器械的架子包装上或医疗器械的外壳上以及在包装医疗器械的每个外壳上印刷以下内容。
1.医疗器械的名称;
2.用户识别设备及其使用所必需的细节;
3.制造商的名称和制造该设备的制造场所的地址;
4.关于净数量的正确说明,应以重量、尺寸、体积、单位数量(视情况而定)以及包装中所含设备的数量以公制表示;
5.生产年月及有效期(或者标签应标明产品的保质期);
6.在需要时提供指示,以表明该器械含有药用或生物物质;
7.提供一个明显的批号,并以“lot no.”一词开头或“lot”或“batch no.”或“b. no.”;
8.在需要时指出适用于该设备的任何特殊存储或处理条件;
9.指明该装置是否以无菌产品形式提供,及其无菌状态和灭菌方法;
10.给予警告或预防措施(如果认为相关的话),以引起医疗器械使用者的注意;
11.如果该设备是一次性使用的,则应适当标记该设备;
12.如果打算将医疗器械作为免费样品分发给医疗专业人员,则在该器械的标签上套印“physician’s sample—not to be sold”字样;
13.除进口设备外,在“manufacturing licence number”或“mfg. lic. no.” 或“m. l”之前加上制造许可证号;
14.如果是进口设备,则在标签上通过粘贴方式提供(如果尚未打印)此类细节,包括:进口许可证编号、进口商的名称和地址、实际生产场所的地址以及制造日期;
15.如果小型医疗器械因为体积原因,无法清晰打印所有信息,则至少应包括产品识别和安全所需的信息。例如:第1、2、3、4条款所涵盖的信息,第5、7、11和13应包括在内。
唯一的器械标识:条例第46条规定,自2022年1月1日起,经批准制造、销售、分销或进口的医疗器械应带有唯一的器械标识,其中应包含器械标识符和生产标识符。其中:
“器械标识符”是指全球贸易项目编号。
“生产标识符”是指序列号、批次或批号、医疗器械软件版本号、制造日期和或有效期。
参考资料:
[1]千羽杂谈:印度对进口医疗设备的依赖“令人担忧”,主要从中国进口!
[2]MedTrend医趋势:印度能否成为全球医疗器械产业链下一站?对中国医疗企业有哪些挑战与机遇?
[3]海关热线:【通关监管】出口印度医疗器械须知.

来源:Internet


