对于植入和III类器械的制造商申请CE认证来说,困扰的首要问题通常都是到底是否需要进行临床试验;若是不做临床试验,走等同路径到底是否一定需要合同?MDCG协调小组似乎洞察到了大家的这一疑问,这次发布的指南文件MDCG2023-7直指这两大敏感问题。本指南旨在澄清将投放欧洲市场的植入式和III类医疗器械的临床试验要求豁免以及与等效性证明相关的相关条件。它还提供了与根据MDR附录XIV第3节证明“足够程度的数据访问”相关的例子和考虑因素。
临床试验要求豁免
对于III类和植入医疗器械来说,MDR原文中提到的4种可豁免进行临床试验的情况:
1.Article 61(4) 前三个短划线所指明的情况:
— 该器械由同一制造商对已投放市场的器械进行改造而成;
— 依据附录XIV第3节规定,已改进的器械经制造商证明后等同于投放市场的器械,且此证明已得到公告机构认可;
— 对投放市场的器械进行临床评价足以证明已改进的器械符合相关的安全性能要求
在这种情况下,公告机构应核查 PMCF(上市后临床跟踪) 计划是合适的,且其中包括上市后研究,以证明器械的安全和能。此外,MDR第61条第 6 段所述的情况无需进行临床研究。
2.Article 61(6)(a):
此类器械依据第 90/385/EEC 号指令或第 93/42/EEC 号指令已合法投放市场或投入使用,其临床评价:
— 基于足够的临床数据;
— 符合此类器械临床评价相关的产品CS(如果有)。
3.Article 61(6)(b):
针对缝线、订书钉、牙齿填充物、牙套、齿冠、螺钉、楔子、板、电线、别针、小夹和连接器而进行的临床评价均应建立在充分的临床数据上且符合相关的产品特定的 CS(如果有)。
4.Article 61(5):
依据MDR第61条第4段规定,制造商生产出的器械经证实等同于已投放市场的器械(不属于同一制造商生产),除此条所要求的内容外,若以下条件均满足,则无需进行临床研究:
— 在这两个制造商拟定合同的适当位置明确允许第二种器械制造商在现有基础上全权使用技术文件。
— 原始临床评价已依照MDR要求执行,且第二种器械制造商向公告机构提供其明确证据。
总结:
对于植入和III类器械来说,若能满足如下其中一条,则有可能豁免进行临床试验:
● 该产品是本公司已经上市产品改型而成,且能证明等同;
● 该产品已获取MDD下的CE认证证书,且基于较为充足的临床数据;
● 该产品为WET清单中的器械;
● 与任何一家可以作为等同的器械的制造商签订了授权合同;
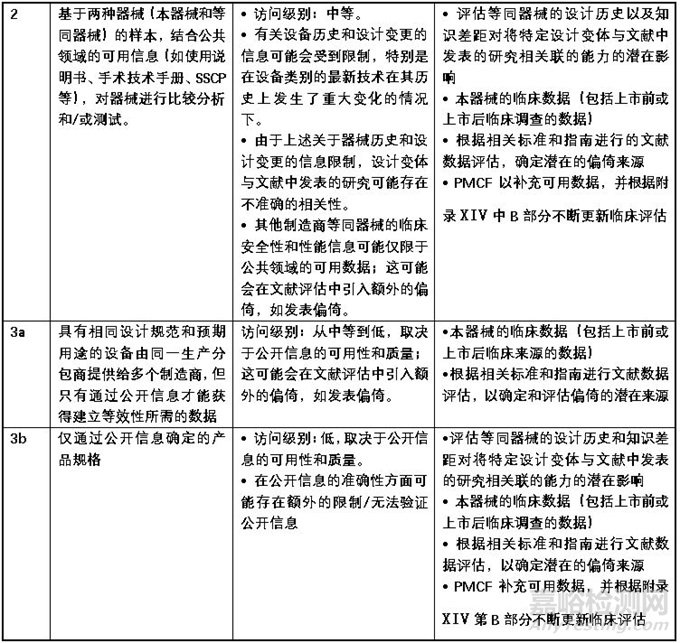
获取数据的充分程度
MDCG2023-7强调,证明“足够的访问水平”并不需要在所有情况下签订合同。合同仅适用于MDR中第61(5)条所述的豁免情况。还应注意的是,MDR的附录XIV第3节专门提到了证明等效性声明所需的数据:即,要求有足够的途径来确定评估等效性所依据的临床、技术和生物特征,而不是获得完整的技术文件。
MDCG2023-7用附表2的形式对“充分访问数据”的方法进行了阐述,该表格内容对公告机构和制造商来说都是评估等同数据是否可认为充分获得的重要参考。