您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-07-06 10:36
摘 要 / Abstract
本文旨在深入探讨美国和欧盟的医疗器械上市后监管制度,包括风险监管理念、监管法规体系、上市后临床研究、质量管理体系等,总结出开发风险监测工具、落实风险管理、采用合并报告模式、公开风险信息等经验,为我国医疗器械行业有序发展提供参考。我国可以借鉴美欧医疗器械上市后的监管实践,从提高监管机构能力、建设监测哨点和强化法规框架等方面,建立和试点医疗器械警戒制度。
This paper aims to explore the post-market regulatory systems for medical devices in the United States and the European Union, including risk regulatory concepts, regulatory legal systems, post-market clinical studies, quality management systems. It summarizes the experiences of developing risk monitoring tools, implementing risk management, merging reporting models, disclosing risk information. The aim is to provide insights and references for the systematic development of China's medical device industry. China can learn from the post-market regulatory practices of medical devices in the United States and the European Union to establish and pilot a robust medical device vigilance system, improving regulatory agency capabilities,building monitoring outposts, and strengthening the legal framework.
关 键 词 / Key words
医疗器械;警戒制度;不良事件监测;风险;试点
medical devices; vigilance system; adverse event monitoring; risk; pilot
2023年9月8日,《十四届全国人大常委会立法规划》明确了医疗器械管理法作为“需要抓紧工作、条件成熟时提请审议的法律草案”,首次被列入立法规划。我国现行的《医疗器械监督管理条例》属于行政法规,在一定程度上制约了制度层面的设计、实施、跨部门联合执法和司法衔接。为进一步促进我国医疗器械行业的升级转型,建议借鉴国际经验,建立具有前瞻性的法律法规体系,完善对产品全生命周期的监管,重点加强安全监管,支持和引导行业的发展[1]。本文基于现行的医疗器械监管法规体系,积极借鉴国内外上市后监管经验,主动顺应医疗器械产业全球化趋势,立足我国国情,积极探索适用于我国的医疗器械监管新模式、新路径,以期推动新工具、新标准、新方法的监管创新[2],助力医疗器械产业高质量发展。
1、医疗器械监管理念
1.1 美国
美国食品药品监督管理局(FDA)下设医疗器械与放射卫生中心(CDRH)通过推进监管科学,为行业提供可预测、一致、透明和高效的监管途径,旨在做到:快速识别性能不佳的医疗器械,帮助患者能够及时和持续地使用高质量、安全和有效的医疗器械;准确描述医疗器械在真实世界的性能,促进设备创新,加速批准或许可;消费者、护理人员和医疗服务提供者基于可访问的医疗器械科普知识,可以作出医疗保健决策。
1.2 欧盟
欧盟委员会的医疗器械协调小组(MDCG)通过建立一个稳健、透明、可预期和可持续的法律框架,确保医疗器械行业的顺利运作发展,以高度保护患者和用户的健康为基础,为医疗器械设定了较高的质量和安全标准,以解决此类产品的共同安全问题,并支持其创新。
1.3 中国
我国的《医疗器械监督管理条例》指出,医疗器械监管需要遵循风险管理、全程管控、科学监管、社会共治的原则[3]。国家药品监督管理局(National Medical Products Administration,NMPA)也在《关于加强医疗器械生产经营分级监管工作的指导意见》中提出,医疗器械生产经营监管应按照“风险分级、科学监管,全面覆盖、动态调整,落实责任、提升效能”的原则开展。NMPA药品评价中心作为承担医疗器械上市后监测的技术机构,将质量方针设定为“科学评价为基础,风险管理为主线,服务患者为中心”。
2、上市后监管法规体系
2.1 美国
FDA建立了较完善的医疗器械上市后法规体系[4]。美国《联邦法规汇编》第21章(21 CFR)803部分“医疗器械报告”详细规定了医疗器械不良事件的收集和上报过程,要求制造商和医疗机构在发现任何与医疗器械相关的问题时立即报告FDA,并出台了“自愿故障摘要报告计划”,允许将已知的不良事件表现进行合并上报。21 CFR 822法规要求制造商对部分Ⅱ、Ⅲ类医疗器械实施强制上市后监测,必须建立有效的监测计划,收集设备使用过程中的数据,并按方案向FDA提交研究报告。根据21 CFR 806法规,制造商发现并报告处理医疗器械相关问题时应主动采取纠正和移除措施。21 CFR 810法规指出,当发现医疗器械存在严重安全问题时,可以由FDA发起并强制执行召回。21 CFR 820法规明确了质量体系的要求,确保制造商在生产经营活动中遵循严格的质量控制。21 CFR 895法规规定了医疗器械被禁止使用的情形。
2012年,FDA在《加强本国医疗器械上市后监测体系》(Strengthening Our National System for Medical Device Postmarket Surveillance)中提出了4项行动以加强医疗器械上市后监测,分别是:建立医疗器械唯一标识(UDI),并促进将其纳入电子健康信息记录[5];促进发展特定产品的患者注册研究;使医疗器械不良事件报告和分析现代化;开发和使用新的证据生成、综合和评估方法[6]。
通过美国政府投入资源和社会各界参与,美国医疗器械不良事件报告整体数量和质量迅速增长。美国制造商和使用机构器械使用经历数据库(MAUDE)数据显示,2022年美国医疗器械不良事件报告达到295.34万份,约为2012年(48.78万份)的6倍(图1)。

2.2 欧盟
根据欧盟关于医疗器械第2017/745号法规(MDR)Article 83部分,制造商必须建立、维护和更新上市后监测体系,积极、系统地收集、记录并分析医疗器械在整个生命周期中的性能、质量和安全相关数据。Article 84要求制造商必须制定详细的上市后监测计划,收集的信息包括监测数据、市场投诉及同类产品风险信息等。Article 85和Article 86要求制造商定期总结上市后监测计划收集的数据,并需要分析结果和结论。Ⅰ类医疗器械制造商应撰写上市后监测报告;Ⅱa、Ⅱb、Ⅲ类医疗器械制造商需要撰写定期安全性更新报告(PSUR),更新安全和临床性能总结文件、风险获益分析结论、上市后临床随访研究(PMCF)结论、销量和使用人数等。Article 87~90法规要求制造商深入分析医疗器械警戒数据,提交严重不良事件和市场安全纠正措施的报告,对非严重事件进行趋势分析和报告,发现潜在问题并采取必要措施来预防或控制。
2.3 中国
我国医疗器械上市后监管法规主要有《医疗器械不良事件监测和再评价管理办法》《医疗器械召回管理办法》《药品医疗器械飞行检查办法》《医疗器械质量抽查检验管理办法》等[7-10]。2018年8月,国家市场监督管理总局、国家卫生健康委员会发布《医疗器械不良事件监测和再评价管理办法》,规定上报单位需要按照可疑即报的原则,在规定时限内进行报告或评价导致或可能导致严重伤害或死亡的个例医疗器械不良事件。该法规还建立了由群体医疗器械不良事件、定期风险评价报告、重点监测、再评价、境外报告等构成的上市后监测工作体系,进一步明确了各方职责,为推动医疗器械不良事件监测工作提供了重要保障[7,11]。2020年7月,NMPA发布《关于进一步加强药品不良反应监测评价体系和能力建设的意见》,提出探索研究医疗器械警戒制度[12]。经过前期基础研究和准备工作,2023年NMPA出台工作方案,在北京、上海、广东、江苏、山东、重庆、海南和贵州8个省市开展医疗器械警戒制度试点工作。
3、上市后临床研究
3.1 美国
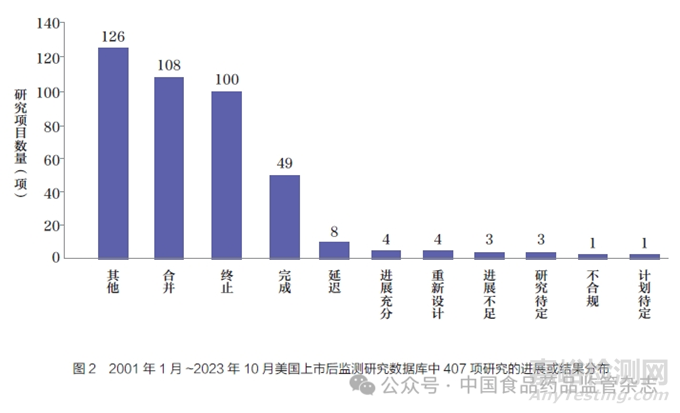
21 CFR 822法规明确了上市后监测的具体范围,包括:医疗器械故障可能造成的严重不良健康后果、植入人体超过1年的医疗器械、在儿童群体中发挥重要作用的医疗器械和院外使用的维持生命类医疗器械等。为了与21 CFR 822法规要求配套,FDA在2022年10月更新了《联邦食品药品和化妆品法案》(Federal Food,Drug,and Cosmetic Act)第522条规定的上市后监测研究(postmarketing surveillance study,PSS)相关指导文件[13]。该文件明确美国制造商需要开展上市后主动监测,对已上市医疗器械产品数据和其他信息进行主动、系统、科学有效地收集、分析和解释。FDA官网公开的PSS数据库在2001年1月~2023年10月共发布347条522研究令,涉及407项研究,研究结果明确为“终止”“重新设计”“不合规”的项目共有105项,占比之和超过25%,产品风险得到有效控制(图2)。除此之外,FDA还建立了由350多家医院组成的医疗器械安全监测网络(Med Sun)、发布了自愿报告系统(Med Watch),成立了国家健康技术评估系统协调中心(NESTcc),发挥了对产品进行上市后监管的重要作用。
3.2 欧盟
根据欧盟MDR法规,制造商有责任进行上市后临床随访(post market clinical follow-up,PMCF)研究,以确保医疗器械的安全有效。这些研究旨在收集有关产品在真实世界使用条件下的效果数据,从而为临床提供更加准确的信息。针对有以下情况的医疗器械,必须开展PMCF研究:创新医疗器械、设计发生变更或预期用途等发生重大变化、产品使用风险较高、针对高风险的解剖部位或高风险的人群、出现了有关安全性和有效性方面新信息和上市前注册通过同品种临床评价路径的产品。欧盟法规明确了开展PMCF研究的主要目的为确认医疗器械在其预期使用寿命内的安全性和性能,识别之前未知的不良事件,监控已识别的不良事件及禁忌症的发生情况,识别并分析突发风险,确保收益/风险比的持续可接受性,确定医疗器械可能的操作不当或超预期使用等。通过这些研究,制造商可以及时发现并解决潜在的安全问题,提高产品的质量和可靠性,同时为医生和患者提供更加安全和有效的治疗选择。
3.3 中国
我国国家药品安全“十二五”规划和“十三五”规划期间,共启动200个医疗器械产品的重点监测工作,首次探索以监测技术机构为主体的主动监测;共建立使用单位监测哨点4561家次,参与重点监测注册人365家次,制定监测方案,监测哨点、相关企业主动参与,收集和分析产品上市后风险信息并综合评价,通过重点监测,共提出无源产品监管建议23条,有源产品监管建议11条,共计对26个产品改进了产品设计和工艺,对19个产品修改完善说明书,增加了警示信息,针对4个产品发布了召回信息或公告,为上市后安全性监管提供技术支撑,针对多个品种加强了使用者沟通和培训,还有一些品种的监测评价结果被转化为国家监管策略的调整内容[14]。“十四五”国家药品安全及促进高质量发展规划期间,37个医疗器械产品重点监测正在开展中[15]。
4、质量管理体系检查
4.1 美国
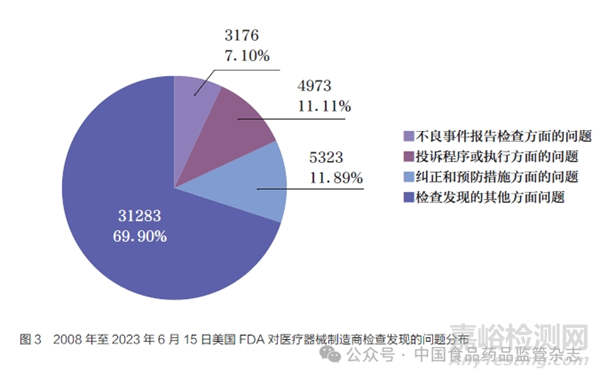
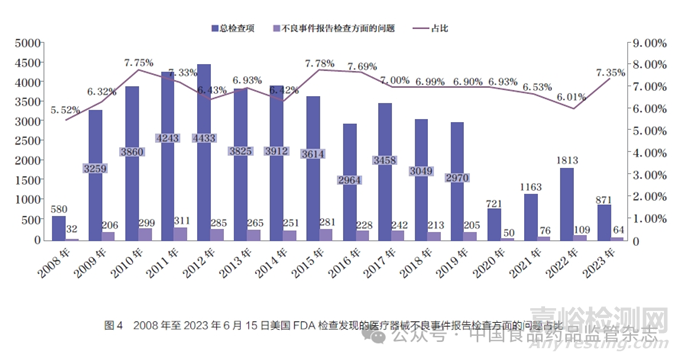
为监督制造商开展上市后医疗器械不良事件报告的落实情况,FDA将21 CFR 803规定的内容列为对制造商开展上市后检查的重要内容(即21 CFR820),并出台了《医疗器械制造商检查指导手册》(Inspection of Medical Device Manufacturers)和《医疗器械PMA产品的上市前批准和上市后检查指导手册》(Medical Device PMA Preapproval and PMA Postmarket Inspections)进行规范。不良事件检查的内容包括制造商收集、评价和报告事件的流程记录及风险识别相关文件。本文分析了2008年至2023年6月15日FDA公开的医疗器械检查问题44 755项,其中纠正和预防措施、投诉程序或执行、不良事件报告检查方面的问题分别占比11.89%、11.11%、7.10%,在所有问题中排名前3,占比之和超过30%(图3)。按年份进行统计发现,不良事件报告检查方面的问题占比均高于5%(图4),表明FDA十分重视医疗器械制造商上市后合规情况。
4.2 欧盟
欧盟开展的医疗器械监督检查旨在确保医疗器械制造商的产品和质量体系运行符合监管要求,遵从医疗器械单一审核程序(medical device single audit program,MDSAP)。MDSAP是以ISO 13485《医疗器械质量管理体系用于法规的要求》(Medical Devices-Quality Management Systems-Requirements for Regulatory Purposes)和美国、澳大利亚、加拿大、巴西、日本5个国家的相关法规为依据,为审核员提供审核方法的指导文件。该文件提供了对医疗器械不良事件和召回报告的具体检查内容,包括:检查制造商是否有识别可能符合参与监管机构定义的报告标准的医疗器械相关事件的流程;检查投诉程序是否有审查每个投诉流程的机制,以确定是否需要向监管机构提交报告;检查制造商的流程是否满足所有出口国家或地区监管机构的时限要求。
4.3 中国
我国于2021年和2022年均开展了医疗器械不良事件监测专项检查,共完成对54家注册人的现场检查,检查共发现不符合项235项,多为一般性缺陷[16]。检查发现的主要问题包括:不良事件监测相关制度或程序文件未及时更新;不良事件收集途径不畅通;不良事件监测相关人员对法规不熟悉;无法提供不良事件监测工作记录;定期风险评价报告未按规范要求撰写或未按法定时限要求提交;产品信息未录入监测系统或未及时更新;严重不良事件报告的调查、分析和评价超出法定时限要求等。以上不符合项对应的相关规定大多属于我国医疗器械上市后监管法规的基本要求。
5、可供我国试点医疗器械警戒制度借鉴的美国和欧盟经验
5.1 开发风险监测工具
美国FDA虽然尚未对医疗器械警戒进行明确定义,但已通过建立开放式数据库(如MAUDE)、医疗器械安全监测网络(Med Sun)、自愿报告系统(Med Watch),探索对医疗器械相关信息的综合分析和共享(如NESTcc等),从而广泛收集医疗器械风险信息。欧盟通过建立医疗器械数据库(EUDAMED),整合了注册许可、医疗器械UDI、公告机构和证书、警戒和上市后监督、临床试验和性能研究、市场监管6个模块,用于整理和处理医疗器械和制造商的信息。可见,美国和欧盟监管机构致力于通过开发上述上市后风险监测工具,构建一个国家级的信息化监测体系,借助受隐私保护的分布式数据系统和通用的数据标准,利用常规收集的包含UDI的电子健康信息进行实时主动监测,加强上市后风险控制,确保医疗器械安全使用。
5.2 落实风险管理
美国FDA和欧盟均要求医疗器械制造商采用系统性的方法来识别、评估和控制风险,并要求其建立和维护有效的风险管理程序,包括明确定义风险和风险控制措施,对设计开发、生产制造和流通使用等过程中的各种风险因素进行详细分析。医疗器械生产企业应针对每个产品制定风险管理计划,使用合适的风险评估工具(如风险矩阵、趋势控制图或风险优先级指南)识别风险、描述风险控制措施和报告机制,以及对风险管理进行验证,还要求具有完备的风险管理文件,包括详细的风险分析、风险评估和风险控制策略,并根据需要进行修订和更新。
5.3 采用合并报告模式
自2018年起,美国FDA出于鼓励和方便医疗器械制造商报告的目的,开始接受故障类不良事件的汇总报告,取代大量故障事件的报告。这项措施为制造商提供了一个新选择,通过在合并报告中增加描述分析的文本,使医疗器械的故障表现更为突出,收集的信息更有价值并更易于理解,同时由于报告和调查不良事件的时限更为宽松,有效减少了需要提交补充报告的频率。
5.4 公开风险信息
美国FDA向社会公众开放共享医疗器械风险信息,任何人可以在告医师函(letter to health care providers)、产品召回数据库(medical device recalls)、安全通讯专栏(safety communications)、撤市禁令专栏(medical device bans)中按照制造商、产品、分类与事件类型等检索条件进行检索。
6、对我国推进医疗器械警戒制度试点的建议
6.1 提高监管机构能力
在医疗器械警戒制度试点期间,建议重点加强监管部门的技术和专业能力,研究先进的监测技术和设备,指导注册人在开展不良事件监测工作的基础上,适当减轻文书负担[17],通过搭建医疗器械警戒体系,撰写警戒计划,主动收集警戒数据,开展风险分析、评估和控制[18-19]。同时,建议研究建立医疗器械警戒信息共享机制,将警戒工作与审评、检查、检验、稽查等工作有机衔接,服务医疗器械全生命周期监管。此外,建议研究建立医疗器械警戒信息跨部门通报渠道,及时共享信息,更好地服务于医保、医疗、医药“三医”协同发展和治理。
6.2 建设监测哨点
建议探索强化哨点医院管理制度,明确参与医疗器械警戒试点的哨点医院工作要求,探索制定哨点医院考核制度,进行动态管理和激励。通过在临床推广患者登记研究、上市后跟踪随访研究等,获取高质量的主动监测数据,将“与使用医疗器械相关的问题”纳入监测范围,进一步扩大监测的覆盖面,丰富风险信号收集范围,提升风险识别能力,弥补自发报告系统在报告完整性、准确性等方面的缺陷。加强监测数据利用度,在不公开注册人信息的情况下,对多维度数据库中的分类产品不良事件进行分析,并将部分产品风险分析数据公开,以便注册人、医疗机构和社会公众获得医疗器械使用经验相关数据。
6.3 强化法规框架
建议明确医疗器械全生命周期监管要求,包括审批前的评估和审批后的监管,推进实施风险管理标准[20-21],提高注册人风险管理能力,推进风险分析和风险管理计划的实施。建议医疗器械注册人依照警戒计划开展上市后研究,或由监管部门依托专业学组和学科带头人发起的安全性评价研究,根据品种特点收集不良事件、投诉或登记研究数据,规范研究过程,获取准确、完整、及时的医疗器械上市后监测数据,提高风险信号识别能力和风险控制能力。
7、展 望
美国和欧盟的医疗器械监管经验可以为我国搭建和试点医疗器械警戒制度提供借鉴。建议从制度、机制、模式和理念上,深入拓展监测工作的广度和深度,强调主动性和广泛性,以我国现有医疗器械不良事件监测工作为基础,进一步落实质量安全主体责任,探索建立医疗器械警戒制度及其运行机制,顺利完成警戒制度试点任务。

来源:中国食品药品监管杂志


