您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-09-27 08:28
溶出接受标准制定
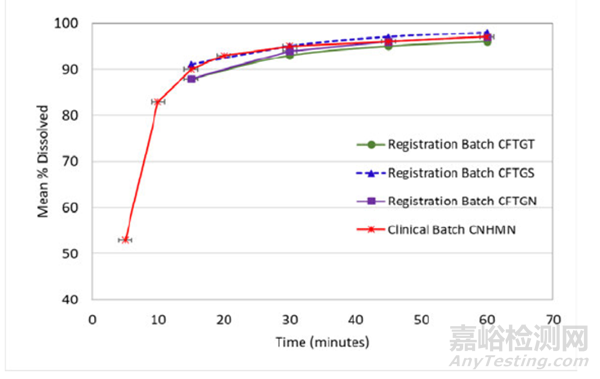
图G:3批注册批次和1批代表性批次的溶出曲线
图G显示了3个注册批次24个月的完整溶出曲数据,以及1个代表性的临床批次溶出数据。
FDA建议,拟定的溶出接受标准从XXmin,XX%收紧为XXmin,XX%。
根据企业提供的资料,图G溶出曲线显示,稳定性样品repotrectinib胶囊的没有变化。基于上述数据,24个月注册批次和代表性临床批次的溶出结果满足收紧后的溶出标准。溶出数据在XXmin的波动较小。
对于低溶解度化合物,API粒度分布被认为是可能影响药物产品性能的关键质量属性。基于上述图D,FDA推荐的溶出接受标准,而不是企业拟定的接受标准,通过2阶段测试可以区分出故意使用原料药粒度D90 XXum制备得到的批次,该粒度值在原料药粒度限度范围外。
而且,对于属于BCS IV类的repotrectinib,添加不同比例的SLS可能会影响repotrectinib胶囊生物利用度。基于上述图E,FDA推荐的溶出接受标准,而不是企业拟定的接受标准,通过2阶段测试可以区分出故意制备无SLS的胶囊批次。
此外,基于临床批次的平均溶出曲线数据(如Figure 3.2.P.5.6-1 and Table 3.2.P.5.4-4),FDA推荐的溶出标准是合适的。
根据拟定的标签,repotrectinib没有表现出QT延长潜力;因此,更快溶出产品批次的安全风险较低。
「解读」针对BCS 4类化合物,溶出区分力考察因此,原料药粒度是必选项,企业也制备得到了一个合适的批次用于确认溶出方法的区分力。另外辅料SLS的比例不同,也可以被溶出方法很好的区分不同比例批次。最后是收紧的溶出接受标准很好的满足了各临床批次和注册批次的放行,以及24个月的稳定性测试。
稳定性批次溶出
根据稳定性数据和ICH Q1E,在USP受控室温条件下存储时,FDA同意拟定有效期为36个月。
根据企业提供的制剂注册批次在长期和加速条件下的数据,24个月都是稳定并没有显示趋势的变化。
而且企业提供的制剂注册批次/临床批次的稳定性数据,通过USP1阶段或2阶段测试,都可以符合修订后的溶出接受标准,无稳定性趋势变化。
由于repotrectinib是一种低溶解度化合物,FDA同意API粒度分布是repotrectinib胶囊的关键生物利用度属性。FDA同意企业理由,尽管拟定的溶出接受标准无法拒绝故意使用大粒度原料药制备得到的批次,但有足够的原料药PSD粒度接受标准,可以用来控制粒度过大而得到的批次,带来溶出不符合要求的风险。因此基于企业提供的数据,溶出接受标准收紧为“20分钟时Q= XX%”。企业接受FDA的这一建议并修订了NDA文件。
针对溶出方法,FDA建议到:
基于企业提供的数据,高温强降的样品与控制样品对比,没有显示出溶出的差异。因此FDA认为,拟定的溶出方法对于高温强降的样品没有区分力,但可检测出现胶囊胶连情况的批次。
拟定的溶出方法没有研究区分具有API多晶型的药物批次的能力。根据企业的研究,原料药另外一种晶型被认为与原料药和制剂生产无关。此外,在SN-15中,申请人提供了原料药确认性XRD和NIR数据,以证明在制剂生产中和加速条件下(在HDPE瓶和开放盘中)长达3个月期间没有观察到API型态变化,因此认为,在药品有效期内没有API晶型变化的风险。
「解读」针对溶出方法和接受标准,最后一点需要评估稳定性批次的溶出数据。首先修订收紧的溶出标准不会让当前注册稳定性批次/代表性批次稳定性失败;其次说明即使溶出方法无法区分超出接受标准大粒度原料药制得的批次,但原料药粒度限度可以控制该风险。FDA进一步评估了拟定的溶出方法无法区分高温强降条件的批次,但可以检测出现胶连胶囊批次样品;另外,拟定的溶出方法无法区分不同原料药晶型得到的批次。
制剂变更桥接
拟定的商业化40mg规格制剂已用于1期、2期和注册稳定性研究,40mg规格的胶囊组成,在临床、注册和拟商业化也是相同的;只有印字胶囊标志不同,该变化仅是外现的非功能性变化,评估不会影响药物溶出和吸收。
所有的临床研究(在模块2.7的表2和表8中){包括TPX-0005-01(TRIDENT-1),TPX-0005-07(儿科研究),研究TPX-0005-08(相对生物利用度/RBA),TPX-0005-09 A部分(绝对BA),TPX-0005-11(食物作用研究),研究TPX-0005-12(RBA),TPX-0005-14(BE和食物效应)}使用了一批或多批的40mg规格制剂,也是拟商业化的规格。在关键临床研究(TRIDENT-1 )中使用了拟商化业工艺处方和拟商业化工艺生产的原料药。
根据表3.2.P.5.4-3(批分析数据)和拟定标签,以及表3.2.P.5.4-2(批量信息),包括印字和非印胶囊,都已用于关键临床研究(TRIDENT-1 )和其它的临床的研究。根据表3.2.P.5.4-2信息和关键临床研究TRIDENT-1 的报告,使用到的批次生产使用的是拟商业化的设施。
而且,用于关键临床的印字与非印字胶囊批次,使用了拟商业化生产商生产,使用了拟商化业工艺和拟商业化工艺生产的原料药,这些批次使用的也是拟商业包装,并进行了稳定性研究。
所有临床批次,包括关键临床批次,展示了非常快速的溶出数据,因此支持了拟商业化的生产规模。
企业承诺会测试3批PPQ批次的溶出数据,用于确认拟定的接受标准。
「解读」变更方面只看到有处方规格和工艺的变更,没有生产厂地的变更,而且企业是在关键临床之前完成了相应的规格和工艺变更,因此拟定的商业化处方40mg规格用在了临床1期,关键临床,注册批次,并将用于商业化的生产。
这种策略可以让企业免除体内的桥接,至多进行体外的桥接。这一信息在企业与FDA的沟通交流中也得到了确认,企业与沟通了规格的变更和溶出方法的变吏,没有提及需要桥接体内的研究,为企业加速上市创造了便利条件,CMC的研究和效率非常高效。

来源:Internet


